LC-MS vs. GC-MS for Natural Product Metabolomics: A Strategic Guide for Method Selection and Integration
This article provides a comprehensive, comparative analysis of liquid chromatography-mass spectrometry (LC-MS) and gas chromatography-mass spectrometry (GC-MS) as core platforms for natural product metabolomics.
LC-MS vs. GC-MS for Natural Product Metabolomics: A Strategic Guide for Method Selection and Integration
Abstract
This article provides a comprehensive, comparative analysis of liquid chromatography-mass spectrometry (LC-MS) and gas chromatography-mass spectrometry (GC-MS) as core platforms for natural product metabolomics. Targeted at researchers, scientists, and drug development professionals, it explores the foundational principles, distinct methodological workflows, and complementary analytical coverages of each technique. The content addresses common troubleshooting and optimization challenges specific to natural product analysis and offers a validated decision-making framework for platform selection based on compound properties and research goals. By synthesizing key insights and highlighting the growing trend toward multiplatform integration, the article aims to empower scientists to design more robust, comprehensive, and impactful metabolomics studies for natural product discovery and characterization.
Core Principles and Historical Evolution: Understanding the GC-MS and LC-MS Platforms
1. Introduction: The "Omics" Lens on Natural Products
The comprehensive study of small molecules, or metabolomics, along with the related discipline of metabonomics which measures dynamic metabolic responses to stimuli, represent powerful approaches in natural product research [1]. These fields aim to characterize the full complement of metabolites—the metabolome—within a biological sample, providing a direct functional readout of physiological or pathological states [1]. For researchers investigating complex natural matrices like plants, fungi, or marine organisms, mass spectrometry (MS) coupled with chromatographic separation has become the cornerstone technology [2]. The central analytical decision often hinges on choosing between Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS), each with distinct operational principles and application scopes. This guide provides a comparative analysis of these platforms, focusing on their performance, experimental protocols, and suitability for different facets of natural product metabolomics.
2. Core Definitions and Conceptual Workflow
- Metabolomics: The comprehensive study of the total measurable metabolite pool in a biological sample under a particular set of conditions. It focuses on the identification and quantification of small molecule metabolites (typically 50-1500 Da) [1].
- Metabonomics: The quantitative measurement of the dynamic, multi-parametric metabolic response of living systems to pathophysiological stimuli or genetic modification. It is more focused on pattern recognition and systemic profiling [1].
A generalized, high-level workflow for mass spectrometry-based metabolomics in natural product research involves several critical, sequential stages [1].
Metabolomics Workflow for Natural Products
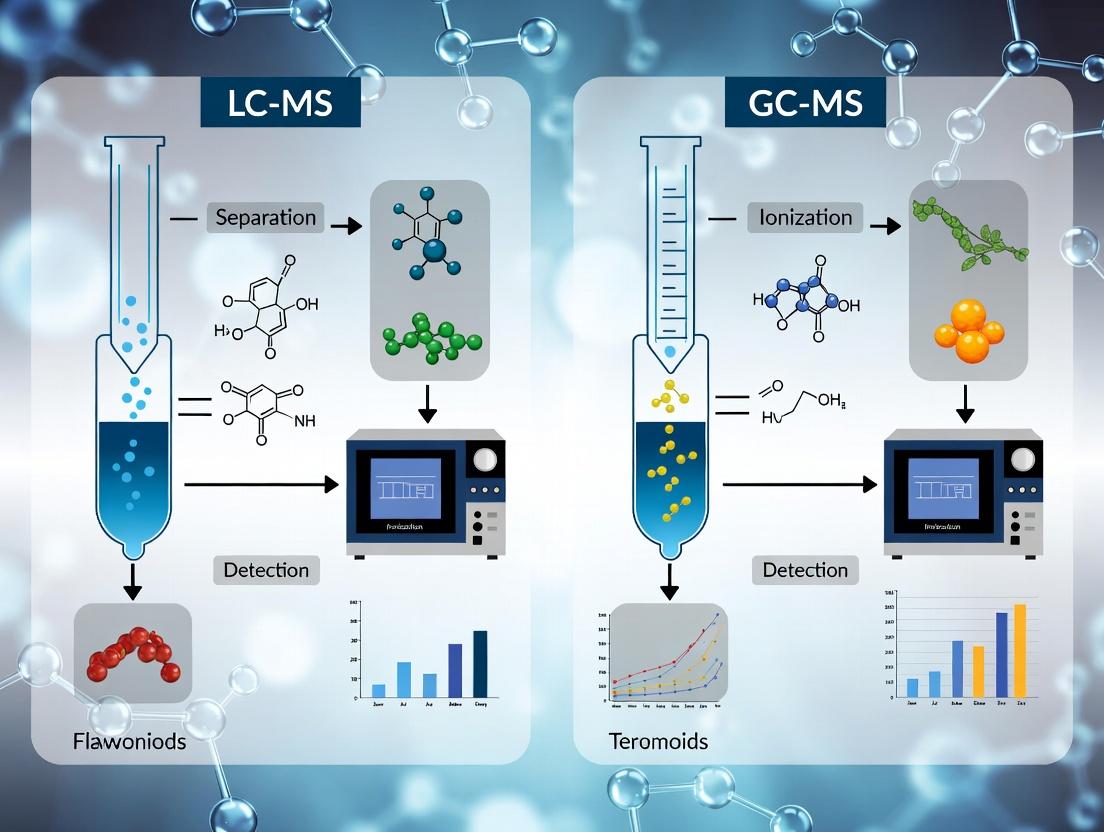
3. Technology Comparison: LC-MS vs. GC-MS
The choice between LC-MS and GC-MS is fundamentally dictated by the physicochemical properties of the target metabolites. The following table summarizes their core operational differences and strengths.
Table 1: Foundational Comparison of LC-MS and GC-MS Platforms
| Feature | Liquid Chromatography-Mass Spectrometry (LC-MS) | Gas Chromatography-Mass Spectrometry (GC-MS) |
|---|---|---|
| Core Principle | Separation in liquid phase (column chemistry), followed by soft ionization (e.g., ESI, APCI). | Separation in gas phase (volatility), followed by hard ionization (Electron Ionization, EI). |
| Ideal Analyte Profile | Non-volatile, thermally labile, polar to semi-polar, and high molecular weight compounds [3]. | Volatile, thermally stable, low molecular weight compounds. Requires derivatization for polar metabolites [4]. |
| Primary Strength | Broad, untargeted coverage of diverse chemical classes without derivatization. Excellent for secondary metabolites [2]. | High chromatographic resolution, superior reproducibility, and robust, searchable spectral libraries for identification [4] [3]. |
| Key Limitation | Matrix effects (ion suppression/enhancement). Less standardized spectral libraries compared to GC-MS. | Limited to volatile or derivatizable compounds. Derivatization adds complexity and can be incomplete [5]. |
| Typical Natural Product Targets | Flavonoids, phenolic acids, alkaloids, saponins, peptides, most lipids. | Terpenes (mono-/sesquiterpenes), essential oils, volatile organic acids, sugars, amino acids (after derivatization). |
4. Performance Comparison with Experimental Data
Empirical studies highlight the complementary nature of these techniques. A multi-omics study on Artemisia argyi Folium used both platforms to achieve comprehensive metabolite profiling [6]. Furthermore, research optimizing GC-MS run times demonstrates practical trade-offs in that technology [7].
Table 2: Quantitative Performance in Natural Product Profiling
| Study & Technique | Key Performance Metrics & Outcomes | Experimental Context |
|---|---|---|
| Multi-omics Profiling of Artemisia argyi [6] | Goal: Systematic metabolite profiling and geographical origin tracing. | |
| • HS-SPME-GC–MS | Identified 66 volatile compounds, primarily monoterpenes and sesquiterpenes. 12 volatile markers differentiated origins. | Headspace Solid-Phase Microextraction (HS-SPME) for volatile capture. |
| • UHPLC-Q-TOF-MS | Identified 32 major non-volatile compounds (e.g., flavonoids, phenolic acids). 12 non-volatile markers differentiated origins. | Reverse-phase chromatography with a Q-TOF mass analyzer. |
| GC-MS Run Time Optimization [7] | Goal: Balance metabolite coverage, reproducibility, and throughput in untargeted GC-MS metabolomics. | |
| • Short Method (26.7 min) | ~138-186 annotated metabolites across matrices. Higher throughput enables full batch analysis within 24h derivatization window. Slightly lower repeatability (RSD ~23-30%). | Compared against a standard 37.5 min method. |
| • Long Method (60 min) | ~175-244 annotated metabolites. Increased coverage from better chromatographic resolution and deconvolution. Better repeatability (RSD ~20-24%). |
5. Detailed Experimental Protocols
5.1. Generic Metabolite Extraction Protocol (Preceding LC-MS or GC-MS) A typical biphasic liquid-liquid extraction protocol for natural product tissues (e.g., plant leaves) is as follows [1] [4]:
- Quenching & Homogenization: Rapidly freeze sample in liquid N₂ and homogenize to a fine powder.
- Solvent Addition: Add a pre-chilled extraction solvent mixture (e.g., methanol:chloroform:water, 2.5:1:1 ratio). Include internal standards (e.g., stable isotope-labeled analogs) for quantification.
- Vortexing & Sonication: Mix vigorously and sonicate in an ice bath for 10-15 minutes.
- Centrifugation: Centrifuge at high speed (e.g., 14,000 g, 15 min, 4°C) to pellet debris and proteins.
- Phase Separation (for biphasic): Transfer supernatant. Adding water/chloroform may induce phase separation, partitioning polar (aqueous) and non-polar (organic) metabolites.
- Concentration: Dry the separated phases under a gentle stream of nitrogen or in a vacuum concentrator.
- Reconstitution: Reconstitute dried extracts in a solvent compatible with the downstream analysis (e.g., LC-MS mobile phase or GC-MS derivatization reagent).
5.2. GC-MS Specific Derivatization Protocol For GC-MS analysis of polar metabolites, a common two-step derivatization is required [4]:
- Methoximation: Dissolve dried extract in methoxyamine hydrochloride in pyridine (e.g., 20 µL, 20 mg/mL). Incubate (e.g., 90 min, 30°C) to protect carbonyl groups by converting ketones and aldehydes to methoximes.
- Silylation: Add a silylating agent like N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) with 1% TMCS (e.g., 80 µL). Incubate (e.g., 60 min, 37°C) to replace active hydrogens in -OH, -COOH, -NH groups with trimethylsilyl groups, increasing volatility and thermal stability.
- Analysis: Analyze derivatized samples typically within 24 hours to prevent degradation [7].
6. The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Materials for Natural Product Metabolomics
| Item | Function & Rationale | Typical Example(s) |
|---|---|---|
| Extraction Solvents | To quench metabolism and solubilize metabolites based on polarity. Choice dictates metabolite coverage. | Methanol (MeOH), Chloroform (CHCl₃), Water (H₂O), Methyl tert-butyl ether (MTBE) [1]. |
| Internal Standards (IS) | To correct for variability in extraction, derivatization, and instrument response for accurate quantification. | Stable isotope-labeled metabolites (¹³C, ²H), e.g., ¹³C-Glucose, D₄-Succinic acid [1]. |
| Derivatization Reagents | (For GC-MS) To chemically modify polar, non-volatile metabolites into volatile, thermally stable derivatives. | Methoxyamine HCl, N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) [4]. |
| Quality Control (QC) Sample | A pooled sample representing all test samples, analyzed repeatedly to monitor instrumental stability and correct for signal drift. | Equal-volume aliquots from every study sample combined into a single QC pool [8]. |
| LC-MS Mobile Phase Additives | To modulate ionization efficiency and chromatographic separation in LC-MS. | Formic acid, Ammonium acetate, Acetonitrile (ACN) [6]. |
| SPME Fiber | (For headspace GC-MS) To adsorb and concentrate volatile organic compounds from sample headspace for sensitive analysis. | Supelco SPME Fiber Assembly (e.g., DVB/CAR/PDMS) [6]. |
7. Data Processing & Analysis Pathways
Following data acquisition, raw spectral data undergoes processing to generate interpretable biological information. For LC-MS, modern unified software pipelines like MetaboAnalystR 4.0 can manage the workflow from raw spectra to functional interpretation [9]. The data analysis logic for a comparative metabolomics study is shown below.
Data Analysis Logic for Comparative Metabolomics
8. Conclusion & Strategic Selection Framework
LC-MS and GC-MS are complementary, not competing, technologies in natural product metabolomics. The decision framework for selecting the appropriate platform depends on the research question:
- Choose GC-MS when: The target compounds are volatile (e.g., essential oils, aroma compounds), or are polar primary metabolites (sugars, organic acids, amino acids) where a robust, standardized, and quantitative workflow with high chromatographic resolution is paramount, and derivatization is acceptable [4] [3].
- Choose LC-MS when: The research requires broad, untargeted coverage of secondary metabolites (flavonoids, alkaloids), analysis of thermally labile or non-volatile compounds, or high-throughput screening with minimal sample preparation [2] [3].
- Employ a Multi-Platform Strategy when: A comprehensive, systems-level view of the metabolome is necessary. As demonstrated, combining HS-SPME-GC–MS for volatiles and UHPLC-MS for non-volatiles provides the most complete chemical portrait of a natural product sample [6].
Ultimately, the scope of metabolomics and metabonomics in natural products is best defined by the strategic integration of these powerful analytical techniques, each illuminating different facets of the complex chemical tapestry produced by living organisms.
Historical Evolution of Metabolic Profiling Technologies
The formal discipline of metabolomics, defined as the global profiling of metabolites in a cell, tissue, or organism, emerged at the end of the 1990s [10]. However, the foundational work that would enable this field began decades earlier with the development of Gas Chromatography-Mass Spectrometry (GC-MS). This technique became one of the earliest and most standardized platforms applied in metabolomics research [4].
The conceptual and technical origins are rooted in the mid-20th century. The principles of gas-liquid partition chromatography were established in the early 1950s [11]. The pivotal advancement came in 1952 with the development of the first GC-MS instrument by J.C. Holmes and F.A. Morrell [12]. This coupling of separation and detection technology created a powerful tool for analyzing complex mixtures. Throughout the 1960s and 1970s, researchers established robust GC-MS protocols for analyzing specific classes of metabolites like sugars, amino acids, sterols, and fatty acids [4]. By the 1970s, scientists were combining targeted analyses of these compound classes into broader profiling assays, a precursor to modern untargeted metabolomics, and even using such profiles to aid in disease diagnosis [4].
In these early years, GC-MS established key advantages that cemented its role. The use of electron ionization (EI) generated reproducible, compound-specific fragmentation patterns [13]. This led to the systematic accumulation of mass spectra in publicly available libraries, most notably the extensive NIST library, which remains a gold standard for identification [4]. The high chromatographic resolution, reproducible retention times, and robust quantification further solidified GC-MS as a foundational technology [13]. While Liquid Chromatography-Mass Spectrometry (LC-MS) later developed with the advent of atmospheric pressure ionization techniques like electrospray ionization (ESI) in the 1980s [12], GC-MS provided the initial model for how separation science coupled with mass spectrometry could deliver a comprehensive chemical profile of a biological system.
Table 1: Historical Milestones in GC-MS and LC-MS Development
| Year | Milestone | Significance for Metabolomics |
|---|---|---|
| 1952 | Development of the first GC-MS instrument [12]. | Enabled the coupling of high-resolution separation with sensitive detection for complex mixtures. |
| 1960s-1970s | Establishment of GC-MS protocols for key metabolite classes (sugars, acids, etc.) [4]. | Created the standardized methods that form the basis for metabolic profiling. |
| 1972 | Emergence of the first LC-MS instrument [12]. | Opened the door for analyzing non-volatile, thermally labile compounds. |
| 1987 | Invention of Atmospheric Pressure Ionization (API) for LC-MS [14]. | Solved critical interface problems, making LC-MS robust and practical for bioanalysis. |
| Late 1990s | Coining of the term "metabolomics" [10]. | Formalized the field of global metabolite profiling, with GC-MS as a core technology. |
| Early 2000s | GC-MS used to identify hundreds of metabolites in Arabidopsis [10]. | Demonstrated the power of the technique for large-scale plant metabolomics. |
Technical Comparison: GC-MS versus LC-MS for Metabolomics
The choice between GC-MS and LC-MS is central to experimental design in natural product research. They are complementary techniques whose operational differences dictate their optimal applications [15].
Core Operational Principles: The fundamental difference lies in the state of the mobile phase. GC-MS uses an inert gas (e.g., helium) to carry vaporized analytes through a heated capillary column, separating compounds based on volatility and interaction with the column coating [12]. In contrast, LC-MS uses a liquid solvent pumped at high pressure through a column packed with fine particles, separating compounds based on polarity, charge, and affinity for the stationary phase [14] [12].
Sample Preparation and Derivatization: This is a major differentiator. Most primary metabolites are not volatile enough for GC. Therefore, GC-MS analysis typically requires a two-step chemical derivatization (methoximation followed by silylation) to reduce polarity and increase thermal stability [13]. LC-MS sample prep is generally simpler but may require careful optimization to remove salts and matrix components that interfere with ionization [15].
Ionization and Identification: GC-MS predominantly uses electron ionization (EI), a "hard" method that produces extensive, reproducible fragment ions. This allows for reliable matching against massive spectral libraries (e.g., NIST, Wiley), enabling confident identification of known compounds [14] [15]. LC-MS primarily uses electrospray ionization (ESI), a "soft" technique that often produces intact molecular ions. Identification relies more on accurate mass, retention time, and MS/MS fragmentation patterns, though library coverage is less comprehensive than for EI [12] [15].
Performance Characteristics: Sensitivity varies by compound class. LC-MS can achieve exceptional sensitivity, down to the 10^-15 mol (femtomole) level for many analytes, making it suitable for trace-level biomarkers [14]. GC-MS sensitivity is also high (10^-12 mol) [14]. GC-MS often provides superior chromatographic resolution, which is particularly advantageous for separating structural isomers [15]. LC-MS, however, covers a much broader range of molecular weights and polarities, from small polar acids to large lipids [12].
Table 2: Direct Comparison of GC-MS and LC-MS Characteristics
| Parameter | GC-MS | LC-MS |
|---|---|---|
| Ideal Analytic Properties | Volatile, thermally stable, typically <500 Da [15]. | Non-volatile, polar, ionic, or thermally labile; broad mass range [14]. |
| Primary Ionization Source | Electron Ionization (EI) [14]. | Electrospray Ionization (ESI) or Atmospheric Pressure Chemical Ionization (APCI) [14]. |
| Typical Identification Method | Matching against extensive EI spectral libraries (NIST, Wiley) [4] [15]. | Accurate mass, MS/MS fragmentation, retention time, smaller libraries [12]. |
| Key Sample Prep Requirement | Often requires chemical derivatization for non-volatile metabolites [13]. | Simpler extraction; may need desalting or specific buffer conditions [15]. |
| Chromatographic Strength | Exceptional resolution for volatile compounds; excellent for isomers [15]. | Can separate a very wide polarity range via different column chemistries [12]. |
| Reported Sensitivity | ~10^-12 mol [14]. | ~10^-15 mol [14]. |
Application in Natural Product Metabolomics: A Comparative Workflow
Natural product research, such as screening medicinal plants for bioactive compounds, benefits immensely from a combined analytical approach. The integrative study of Origanum ramonense provides a clear workflow [16].
Experimental Protocol for Integrative Profiling:
- Sample Extraction: Plant material is extracted with multiple solvents of varying polarity (e.g., methanol, ethyl acetate, hot water) to capture diverse metabolites [16].
- Parallel Analysis:
- GC-MS Analysis: Extracts are derivatized (e.g., via trimethylsilylation) and analyzed. The high separation power of GC resolves volatile compounds and derivatized polar metabolites like organic acids and sugars [16] [4].
- LC-MS Analysis: Underivatized extracts are analyzed, typically using reversed-phase (RP) chromatography to capture mid- to non-polar compounds (e.g., many flavonoids, alkaloids) and possibly hydrophilic interaction liquid chromatography (HILIC) for polar compounds [12].
- NMR Spectroscopy: Provides complementary, quantitative structural information on major constituents and helps identify functional groups without the need for separation [16].
- Data Integration: Metabolite lists from each platform are combined. Multivariate statistical analysis (e.g., Principal Component Analysis - PCA) is used to correlate solvent extracts, identified metabolites, and results from bioactivity assays (e.g., antioxidant, antibacterial tests) [16].
Complementary Coverage: The synergy is evident. A study on Cajanus scarabaeoides seeds used GC-MS to identify 135 volatile metabolites and LC-MS to identify 117 non-volatile metabolites, including flavonoids and polyphenols [17]. GC-MS excels for essential oils, short-chain fatty acids, and primary metabolites, while LC-MS is indispensable for higher molecular weight secondary metabolites like glycosylated flavonoids and thermolabile compounds.
Diagram: Complementary GC-MS and LC-MS Workflow for Natural Product Profiling. The workflow shows how plant extracts are split for parallel analysis. GC-MS requires a derivatization step for polar metabolites, while LC-MS analyzes compounds in their native state. Data from both platforms are integrated for a comprehensive profile.
Selecting the appropriate platform depends on the specific research question and the physicochemical properties of the target metabolites [15].
Strategic Selection Guide:
- Choose GC-MS when: The focus is on volatile compounds (e.g., essential oils, aroma compounds), primary metabolism (organic acids, sugars, amino acids via derivatization), or thermally stable compounds below ~500 Da. It is preferred when confident identification via spectral libraries is critical and for applications requiring high-resolution separation of isomers [4] [15].
- Choose LC-MS when: The target analytes are non-volatile, polar, or thermally labile, such as most secondary metabolites (flavonoids, tannins, alkaloids), peptides, or lipids. It is essential for high-sensitivity targeted quantification of specific biomarkers in complex matrices like plasma [14] [12].
- Employ an Integrated Approach: For comprehensive, untargeted profiling of natural products—where the chemical diversity is immense—using both GC-MS and LC-MS is the most powerful strategy. This multi-platform approach maximizes metabolite coverage and provides stronger evidence for identification [16] [17].
The Scientist's Toolkit: Essential Reagent Solutions
- Derivatization Reagents (for GC-MS): Methoxyamine hydrochloride and N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA). These are used in the standard two-step derivatization to protect carbonyl groups and silylate polar functional groups (-OH, -COOH, -NH), rendering metabolites volatile for GC analysis [4] [13].
- Extraction Solvent Systems: Ternary solvent mixtures (e.g., methanol/acetonitrile/water or isopropanol/acetonitrile/water). These are designed to simultaneously extract a wide range of polar and mid-polar metabolites while precipitating proteins, providing a more comprehensive metabolome coverage than single solvents [4] [18].
- Internal Standards (Isotope-Labeled): Compounds like 13C-labeled amino acids or deuterated lipids. They are added at the beginning of sample preparation to correct for losses during extraction, derivatization, and matrix effects during MS analysis, ensuring accurate quantification [4].
- Quality Control (QC) Pooled Sample: A sample created by combining a small aliquot of every experimental sample. It is analyzed repeatedly throughout the instrumental sequence to monitor and correct for system performance drift over time, ensuring data stability in large-scale studies [4].
Conclusion: GC-MS holds a historic and continuing vital role in metabolomics, offering unparalleled reproducibility, robust identification, and quantitative rigor for volatile and derivatizable metabolites. LC-MS expanded the detectable chemical space to include non-volatile and labile compounds with exceptional sensitivity. For natural product metabolomics, which seeks a holistic view of complex chemical mixtures, the two techniques are not competitors but essential partners. The most insightful research strategies will continue to leverage their complementary strengths within an integrated analytical framework.
Diagram: Decision Framework for Selecting GC-MS or LC-MS. This flowchart guides researchers through key questions about their analytes and goals to recommend the most suitable analytical platform or strategy.
The field of natural product metabolomics seeks to comprehensively identify and quantify the diverse small molecules within biological systems to discover novel therapeutics and understand complex biosynthetic pathways [19] [20]. For decades, analytical chemists have relied on two principal chromatographic techniques coupled with mass spectrometry: Gas Chromatography-Mass Spectrometry (GC-MS) and Liquid Chromatography-Mass Spectrometry (LC-MS). While both are indispensable, their fundamental principles dictate distinct analytical niches.
GC-MS excels in the separation and analysis of volatile and thermally stable compounds. Its historical roots are deep, with some of the earliest "metabolite profiling" research conducted using this platform [20]. However, its requirement for analyte volatility often necessitates chemical derivatization—an extra step that adds complexity, can lead to analyte loss, and is unsuitable for many labile, high-molecular-weight, or polar natural products [21].
In contrast, LC-MS has risen to prominence as the cornerstone of modern metabolomics due to its unparalleled versatility [2] [1]. It directly analyzes compounds in their liquid phase, making it uniquely suited for a vast range of non-volatile, thermally labile, and polar metabolites—characteristics common to many crucial natural product classes like flavonoids, alkaloids, glycosides, and peptides [2] [22]. The development of soft ionization techniques, most notably electrospray ionization (ESI), was a pivotal innovation that enabled the efficient transfer of these delicate molecules into the gas phase for mass analysis without fragmentation, thereby revolutionizing the study of complex biological mixtures [2] [20].
This guide provides a comparative analysis of LC-MS and GC-MS within the context of natural product metabolomics. It objectively evaluates their performance, supported by experimental data and detailed protocols, to inform researchers and drug development professionals in selecting the optimal platform for their specific analytical challenges.
Head-to-Head Comparison: LC-MS vs. GC-MS
The choice between LC-MS and GC-MS is fundamental and hinges on the chemical space of interest. The following tables provide a structured comparison of their core characteristics, performance, and suitability for natural product research.
Table 1: Fundamental Analytical Scope and Technical Requirements
| Aspect | Liquid Chromatography-Mass Spectrometry (LC-MS) | Gas Chromatography-Mass Spectrometry (GC-MS) |
|---|---|---|
| Optimal Analytic Range | Non-volatile, thermally unstable, polar to moderately non-polar compounds (e.g., peptides, lipids, most plant secondary metabolites) [2] [21]. | Volatile and thermally stable compounds; can be extended to polar compounds via derivatization [21] [20]. |
| Typical Ionization Source | Electrospray Ionization (ESI), Atmospheric Pressure Chemical Ionization (APCI) [2] [22]. | Electron Ionization (EI), Chemical Ionization (CI). |
| Sample Preparation | Can be minimal (e.g., protein precipitation, filtration). Extraction tailored to metabolite polarity [1] [21]. | Often requires derivatization (e.g., silylation, methylation) for non-volatile analytes, adding steps and potential for error [21]. |
| Key Strengths | Broad, untargeted coverage of diverse chemistries; analysis of intact, labile molecules; high sensitivity for polar compounds; compatibility with aqueous samples [2] [20]. | Excellent chromatographic resolution and reproducibility; powerful, standardized EI spectral libraries for confident identification; robust and cost-effective [8] [20]. |
| Primary Limitations | Can struggle with very non-polar compounds (e.g., some hydrocarbons); matrix effects can suppress ionization; less standardized universal libraries than GC-MS [21] [9]. | Limited to volatile/derivatizable compounds; high-temperature analysis destroys or alters thermally labile molecules [21]. |
Table 2: Performance in Natural Product Metabolomics
| Performance Metric | LC-MS | GC-MS | Context for Natural Product Research |
|---|---|---|---|
| Metabolite Coverage | Very High. Can detect thousands of features from diverse classes (alkaloids, phenolics, terpenoids, saccharides) in a single run [1] [20]. | Moderate/Limited. Excellent for primary metabolites (organic acids, sugars, amino acids post-derivatization), volatile oils, and fatty acids [21] [20]. | LC-MS is essential for the broad, untargeted discovery of novel secondary metabolites. GC-MS is powerful for targeted profiling of core metabolic pathways. |
| Sensitivity | High (fg-pg level). Advanced instrumentation enables trace analysis in complex matrices like plant extracts [2] [22]. | High (pg level). Highly sensitive for suitable analytes [20]. | Both are highly sensitive, but LC-MS maintains this sensitivity for a wider range of relevant natural product structures. |
| Structural Elucidation | Advanced via tandem MS (MS/MS). Provides molecular weight and fragment patterns. Relies on growing but incomplete databases [20] [9]. | Standardized via EI spectra. Produces reproducible, library-searchable fragmentation patterns [20]. | GC-MS offers more confident identifications for known compounds in its domain. LC-MS/MS is more versatile for unknown de novo structural characterization. |
| Throughput & Robustness | High. Modern UHPLC-MS runs can be under 5 minutes. Robust but requires careful management of ionization suppression [2] [21]. | High. GC offers fast, highly reproducible separations. Long-term signal drift can be an issue but is correctable [8]. | Both are suitable for high-throughput. LC-MS has faster sample prep (no derivatization). GC-MS may require more frequent calibration for quantitation over long periods. |
Table 3: Workflow and Practical Considerations
| Consideration | LC-MS Workflow | GC-MS Workflow | Implication for Research |
|---|---|---|---|
| Sample Preparation | Extraction (e.g., MeOH/CHCl₃, MeOH/H₂O), centrifugation, possibly SPE. Focuses on quenching metabolism and efficient recovery [1]. | Extraction, followed by drying and derivatization (e.g., with MSTFA). Critical for making polar compounds volatile [21]. | LC-MS prep is generally faster and preserves native structures. GC-MS prep is more time-consuming and chemically alters analytes. |
| Chromatography | RPLC (C18) for mid-nonpolar compounds; HILIC for polar compounds. Choice dramatically affects coverage [21] [22]. | Capillary columns with non-polar (e.g., 5% phenyl polysiloxane) or polar stationary phases. Exceptional peak capacity [20]. | LC-MS offers orthogonal separation mechanisms (RPLC vs. HILIC) to expand coverage. GC-MS provides superior resolution for volatile mixtures. |
| Data Analysis Complexity | High. Complex data requires sophisticated processing for peak picking, alignment, and annotation against custom or public MS/MS libraries [1] [9]. | Moderate. Well-established processing pipelines. Mature, universal EI libraries (e.g., NIST) simplify compound identification [8] [20]. | LC-MS data analysis is a major bottleneck but offers greater discovery potential. GC-MS data is more straightforward to interpret for known compounds. |
| Best-Suited Applications | Untargeted metabolomics, natural product discovery, lipidomics, analysis of glycosides, polyphenols, peptides [19] [22]. | Targeted metabolomics of primary metabolism, volatile profiling (essential oils), steroid analysis, environmental metabolite analysis [21] [20]. | The techniques are complementary. LC-MS is the tool for broad discovery, while GC-MS is ideal for specific, volatile-focused, or highly quantitative targeted studies. |
Experimental Protocols for Comparative Metabolomics
To illustrate the practical differences, here are generalized standard operating procedures for untargeted plant metabolomics using both platforms.
Protocol: Untargeted Plant Metabolite Profiling by LC-MS
This protocol is designed for broad coverage of secondary metabolites from plant tissue [1] [22].
Sample Quenching and Homogenization:
- Fresh plant tissue (e.g., 50 mg) is rapidly flash-frozen in liquid nitrogen and ground to a fine powder using a chilled mortar and pestle or a ball mill. This step quenches enzymatic activity to preserve the metabolic profile [1].
Metabolite Extraction:
- The powdered tissue is transferred to a microcentrifuge tube.
- A pre-cooled extraction solvent is added. For comprehensive coverage, a biphasic system is recommended: 1 mL of methanol:chloroform:water (2.5:1:1, v/v/v) [1].
- Internal standards (e.g., stable isotope-labeled analogs of key metabolites) are spiked in at this stage to correct for technical variation [1].
- The mixture is vortexed vigorously for 1 minute, sonicated in an ice-water bath for 15 minutes, and then incubated at -20°C for 1 hour to precipitate proteins.
- Centrifugation at 14,000 x g for 15 minutes at 4°C separates the phases. The upper polar phase (methanol/water, containing polar metabolites) and the lower non-polar phase (chloroform, containing lipids) are carefully collected into separate vials.
- The extracts are dried under a gentle stream of nitrogen or in a vacuum concentrator and reconstituted in 100 µL of initial LC mobile phase (e.g., 95% solvent A: 5% solvent B).
LC-MS Analysis:
- Chromatography: Use a reversed-phase (e.g., C18) UHPLC column for mid-polar to non-polar metabolites. For full coverage, analyze the polar reconstituted extract also via a HILIC column [21] [22]. A typical gradient runs from 5% to 95% organic solvent (acetonitrile or methanol) in water over 10-20 minutes, both acidified with 0.1% formic acid.
- Mass Spectrometry: Operate the mass spectrometer in data-dependent acquisition (DDA) mode. A full MS1 scan (e.g., m/z 70-1200) is followed by MS2 fragmentation scans of the most intense precursor ions. Use electrospray ionization in both positive and negative modes in separate runs to maximize metabolite detection [2] [9].
Data Processing:
- Use computational workflows like those in MetaboAnalystR 4.0 or similar tools (XCMS, MS-DIAL) for peak detection, alignment across samples, and deisotoping [9].
- Annotate metabolites by matching accurate mass (MS1), retention time (if standards are available), and MS2 fragmentation patterns against public spectral libraries (e.g., GNPS, MassBank) [9].
Protocol: Targeted Primary Metabolite Analysis by GC-MS
This protocol is optimized for the quantitative analysis of polar primary metabolites (e.g., organic acids, amino acids, sugars) after derivatization [21] [20].
Sample Extraction for GC-MS:
- Homogenize and quench tissue as in Step 1 of the LC-MS protocol.
- Extract metabolites with 1 mL of a suitable solvent, such as a cold mixture of methanol:water (70:30, v/v).
- Add internal standards (e.g., succinic acid-d4, alanine-d4).
- Vortex, sonicate, centrifuge, and collect the supernatant.
Derivatization (Critical Step):
- Dry an aliquot of the supernatant (e.g., 50 µL) completely in a vacuum concentrator.
- Add 50 µL of methoxyamine hydrochloride (20 mg/mL in pyridine) to protect carbonyl groups. Incubate at 30°C for 90 minutes with shaking.
- Add 100 µL of N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) to silylate hydroxyl and amine groups. Incubate at 37°C for 30-60 minutes.
- Centrifuge and transfer the derivatized supernatant to a GC vial for analysis.
GC-MS Analysis:
- Chromatography: Use a non-polar capillary column (e.g., 5% phenyl polysiloxane). Employ a temperature gradient, typically starting at 60°C, ramping to 330°C.
- Mass Spectrometry: Use electron ionization (EI) at 70 eV. Operate in selected ion monitoring (SIM) mode for highest sensitivity in targeted analysis, or full scan mode (e.g., m/z 50-600) for broader profiling.
Data Processing and Quantification:
- Integrate peaks for target ions. Identify compounds by matching retention times and full EI spectra to commercial libraries (e.g., NIST).
- Quantify using calibration curves built from authentic standards processed through the same derivatization procedure, normalized to the internal standards.
Visualizing Metabolomics Workflows
The following diagrams illustrate the core workflows and decision points for LC-MS and GC-MS in natural product metabolomics.
Diagram 1: Comprehensive LC-MS metabolomics workflow for natural products, highlighting steps from sample preparation to biological interpretation [1] [22] [9].
Diagram 2: Decision tree for selecting between GC-MS and LC-MS based on analyte properties and research objectives [21] [20].
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Research Reagent Solutions for LC-MS and GC-MS Metabolomics
| Item | Function in Workflow | Key Consideration |
|---|---|---|
| Methanol, Acetonitrile (LC-MS Grade) | Primary organic solvents for LC mobile phases and metabolite extraction. Minimize ion suppression and background noise in MS detection [1] [22]. | Must be ultra-pure (LC-MS grade) to prevent contamination and signal interference. |
| Chloroform, Methyl tert-Butyl Ether (MTBE) | Non-polar solvents for lipid and non-polar metabolite extraction in biphasic systems [1]. | Enables comprehensive extraction covering a wide polarity range. |
| Formic Acid, Ammonium Acetate/Formate | Mobile phase additives for LC. Acidifiers improve [M+H]+ ionization in positive mode; buffers aid [M-H]- in negative mode and control retention [21]. | Concentration (typically 0.1%) is critical for consistent ionization efficiency and chromatographic shape. |
| Methoxyamine Hydrochloride | Derivatization reagent for GC-MS. Converts carbonyl groups (in ketones, aldehydes) to methoximes, preventing cyclization and stabilizing sugars [21] [20]. | Essential first step in two-stage GC derivatization to ensure accurate representation of certain metabolite classes. |
| N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) | Silylation reagent for GC-MS. Replaces active hydrogens in -OH, -COOH, -NH groups with trimethylsilyl groups, rendering metabolites volatile and thermally stable [20]. | Makes polar metabolites amenable to GC analysis. Must be kept anhydrous. |
| Stable Isotope-Labeled Internal Standards (e.g., 13C, 15N, 2H) | Added at extraction to correct for variability in sample preparation, injection, and ionization efficiency. Critical for reliable quantification [1]. | Should be chosen to represent different metabolite classes and added at the earliest possible step. |
| Quality Control (QC) Pool Sample | A pooled aliquot of all experimental samples. Injected repeatedly throughout the analytical sequence to monitor instrument stability, align features, and correct for signal drift [8] [9]. | Fundamental for ensuring data quality in large-scale untargeted studies, especially for LC-MS. |
The rise of LC-MS as the preeminent platform for natural product metabolomics is firmly grounded in its ability to analyze the broader, more chemically diverse spectrum of metabolites inherent to biological systems. While GC-MS remains unparalleled for specific volatile and targeted applications, LC-MS's compatibility with labile, high-molecular-weight, and polar compounds aligns perfectly with the needs of modern drug discovery from natural sources [19] [22].
The future of the field lies not in the supremacy of one technique over the other, but in their strategic integration and the continued advancement of LC-MS technology. Emerging trends include:
- Multi-platform Metabolomics: Combining GC-MS data (for primary metabolism) with LC-MS data (for secondary metabolism) to build a more complete biochemical picture of an organism [19] [20].
- Advanced Informatics: Tools like MetaboAnalystR 4.0 are unifying the complex data processing pipeline, from raw spectra to functional insight, addressing a major bottleneck [9].
- Advanced MS and Chromatography: The use of ion mobility spectrometry (IMS) for added separation dimension, and the refinement of hydrophilic interaction liquid chromatography (HILIC) to better capture the polar metabolome, are continually expanding LC-MS's coverage [2] [21].
For researchers embarking on natural product metabolomics, the guiding principle should be fit-for-purpose selection. LC-MS is the undeniable engine for untargeted discovery across vast chemical space. However, a well-designed study may leverage the quantitative rigor and robust identifications of GC-MS for specific pathways, thereby harnessing the complementary strengths of both foundational techniques.
Metabolomics, the comprehensive study of small-molecule metabolites within a biological system, relies fundamentally on analytical techniques capable of separating, identifying, and quantifying chemically diverse compounds [1]. The hyphenated techniques of Gas Chromatography-Mass Spectrometry (GC-MS) and Liquid Chromatography-Mass Spectrometry (LC-MS) form the cornerstone of modern metabolomics research [14]. These systems synergistically combine the superior separation power of chromatography with the sensitive and specific detection of mass spectrometry. The choice between GC-MS and LC-MS is not a matter of superiority but of chemical compatibility, dictated by the physical and chemical properties of the target metabolome [15]. This guide provides a detailed, objective comparison of both platforms, focusing on their operational principles, performance metrics, and optimal applications within natural product and biomedical research.
Fundamental Principles and Instrumentation
Core Components of a GC-MS System
A GC-MS system separates components using a gas chromatograph before ionizing and detecting them with a mass spectrometer [23].
- The Gas Chromatograph: The sample is vaporized in a heated inlet and carried by an inert carrier gas (e.g., helium, hydrogen) through a long, thin capillary column coated with a stationary phase [24]. Compounds separate based on their boiling points and affinities for the stationary phase, eluting at specific retention times [23].
- The Mass Spectrometer: Eluted compounds enter the ion source. The most common method is electron ionization (EI), where a filament emits high-energy electrons (typically 70 eV) that bombard molecules, causing them to fragment into characteristic positively charged ions [23]. These ions are then filtered by a mass analyzer (often a quadrupole) and detected [23]. The resulting mass spectrum provides a reproducible "fingerprint" for compound identification [4].
Core Components of an LC-MS System
An LC-MS system separates compounds in a liquid phase before ionization and mass analysis [25].
- The Liquid Chromatograph: The sample in a liquid solvent is pumped at high pressure through a column packed with a stationary phase [25]. In the common reversed-phase mode, separation occurs based on the hydrophobicity of the analytes [25].
- The Interface and Mass Spectrometer: The central challenge is coupling the liquid stream to the high-vacuum mass spectrometer. This is solved by atmospheric pressure ionization (API) interfaces [26]. The most prevalent is electrospray ionization (ESI), where the LC eluent is nebulized into a fine spray in a strong electric field, creating gas-phase ions from solution [25]. This is a "soft" ionization technique that often produces intact molecular ions with little fragmentation, in contrast to EI [15]. The ions are then guided into the mass analyzer.
Visual Comparison: GC-MS vs. LC-MS Workflow
The following diagram illustrates the fundamental differences in the analytical workflow between the two platforms.
Comparative Performance in Metabolomics Research
Analytical Scope and Compound Coverage
The primary factor dictating platform choice is the inherent chemical nature of the target analytes.
Table 1: Analytical Scope and Compound Class Coverage
| Criterion | GC-MS | LC-MS |
|---|---|---|
| Ideal Molecular Weight | Typically < 650 Da [4] | Broad range, from small molecules to large peptides/proteins (>10 kDa) [15] |
| Key Analyte Properties | Volatile and thermally stable [15]. Non-volatile compounds require chemical derivatization [4]. | Polar, ionic, thermally labile, and non-volatile compounds [15]. |
| Exemplary Metabolite Classes | Organic acids, sugars, fatty acids, sterols, alcohols, amino acids (after derivatization), volatile organic compounds (VOCs) [4] [24]. | Lipids, amino acids (underivatized), flavonoids, nucleotides, peptides, bile acids, glycosides [14]. |
| Ionization Technique | Electron Ionization (EI) [23]. | Electrospray Ionization (ESI) or Atmospheric Pressure Chemical Ionization (APCI) [25] [14]. |
| Typical Ionization Outcome | "Hard" ionization: Extensive, reproducible fragmentation for library matching [23]. | "Soft" ionization: Often produces intact molecular ions ([M+H]⁺, [M-H]⁻); fragmentation requires tandem MS (MS/MS) [25]. |
Performance Metrics: Sensitivity, Identification, and Throughput
Quantitative performance varies based on analyte compatibility and instrument configuration.
Table 2: Performance Metrics and Practical Considerations
| Metric | GC-MS | LC-MS | Experimental Context & Notes |
|---|---|---|---|
| Typical Sensitivity | ~10⁻¹² mol [14]. | Can reach ~10⁻¹⁵ mol [14]. | LC-MS sensitivity is often superior for polar biomolecules in targeted bioanalysis. GC-MS offers high sensitivity for suitable volatile targets [15]. |
| Compound Identification | Excellent via spectral libraries. Large, curated EI libraries (e.g., NIST) enable high-confidence matching [4]. | Relies more on accurate mass and MS/MS fragmentation. Library coverage is growing but less mature than EI libraries [15]. | The NIST 2014 library contains ~242,000 unique compound spectra for GC-MS vs. ~8,000 for LC-MS/MS [4]. |
| Chromatographic Resolution | Very high due to long, efficient capillary columns [15]. | High, but generally less than GC. Advanced modes (e.g., dual-column 2D-LC) improve coverage [27]. | GC is exceptional at separating structural isomers [15]. |
| Sample Preparation | Often complex, requiring derivatization for many metabolites (adds time, variability) [4] [15]. | Usually simpler (dilution, protein precipitation, extraction). Critical control of pH/buffers for ionization efficiency [15] [1]. | Derivatization for GC-MS makes compounds volatile and thermally stable [4]. |
| Operational Cost | Generally lower. Mobile phase is inert gas; maintenance can be simpler [15]. | Generally higher. Costs for high-purity solvents and disposal; potentially more maintenance [15]. |
Detailed Experimental Protocols
This protocol is designed for comprehensive profiling of primary metabolism.
- Sample Quenching & Homogenization: Rapidly freeze tissue in liquid N₂. Homogenize the frozen tissue in a pre-chilled mixture of acetonitrile:isopropanol:water (e.g., 3:3:2) using a bead mill or homogenizer at 4°C. This ternary solvent system aims for broad metabolite coverage [4].
- Lipid Clean-up: Centrifuge the extract. Transfer the supernatant and evaporate to dryness under a gentle nitrogen stream or vacuum. Redissolve the dried extract in a cold methanol-water mixture. Incubate at -20°C to precipitate remaining lipids, then centrifuge and collect the supernatant for drying again [4]. This step is crucial to prevent lipid accumulation in the GC system.
- Chemical Derivatization:
- Methoximation: Dissolve the dried extract in methoxyamine hydrochloride in pyridine (e.g., 20 mg/mL). Incubate (e.g., 90 min at 30°C) to protect carbonyl groups (in sugars, keto acids).
- Trimethylsilylation: Add a silylating agent like N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) with a catalyst (e.g., 1% TMCS). Incubate (e.g., 60 min at 37°C) to replace active hydrogens in -OH, -COOH, -NH groups with trimethylsilyl groups, rendering metabolites volatile [4].
- GC-MS Analysis: Inject 1 µL of the derivatized sample in split or splitless mode. Use a temperature-programmed oven (e.g., 60°C to 330°C) on a mid-polarity stationary phase column (e.g., 5% phenyl polysiloxane). Operate the mass spectrometer in full-scan mode (e.g., m/z 50-600) with electron ionization at 70 eV [4].
- Quality Control: Include pooled "quality control" samples from all study samples, process blanks, and reference standard mixtures throughout the batch run.
This protocol is suitable for untargeted profiling of plasma or serum.
- Protein Precipitation & Metabolite Extraction: Thaw plasma/serum samples on ice. Aliquot a precise volume (e.g., 50 µL) into a cold microcentrifuge tube. Add a chilled organic solvent (e.g., 200 µL of methanol:acetonitrile, 1:1) containing isotopically labeled internal standards for various metabolite classes. Vortex vigorously and incubate at -20°C for 1 hour to ensure complete protein precipitation [1].
- Pellet Removal: Centrifuge at high speed (e.g., 14,000-18,000 x g) for 15-20 minutes at 4°C. Carefully transfer the clear supernatant containing the metabolites to a new vial.
- Sample Reconstitution: Evaporate the supernatant to complete dryness using a vacuum concentrator. Reconstitute the dried metabolites in a volume of starting mobile phase compatible with the LC system (e.g., 100 µL of 98% solvent A / 2% solvent B). Vortex and centrifuge before transfer to an LC vial.
- LC-MS Analysis:
- Chromatography: Utilize a reversed-phase column (e.g., C18). A common gradient employs water (with 0.1% formic acid) and acetonitrile (with 0.1% formic acid) to separate compounds by hydrophobicity [25]. For broader coverage, implement a dual-column setup (e.g., RP and HILIC) to capture both polar and non-polar metabolites in a single run [27].
- Mass Spectrometry: Use electrospray ionization in both positive and negative ion modes. Acquire data in data-dependent acquisition (DDA) mode: a full-scan high-resolution mass spectrum (e.g., m/z 70-1050) is followed by MS/MS scans on the most intense ions for structural elucidation [1].
- Quality Control: Analyze a pooled QC sample repeatedly at the beginning, throughout, and at the end of the batch to monitor instrument stability.
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 3: Key Reagents and Consumables for Metabolomics Sample Preparation
| Item | Primary Function | Platform Relevance | Key Notes |
|---|---|---|---|
| Methoxyamine Hydrochloride | Methoximation reagent; protects keto and aldehyde groups to prevent multiple derivative forms in GC [4]. | GC-MS | Critical for stabilizing sugars and related carbonyl-containing metabolites prior to silylation. |
| N-Methyl-N-(trimethylsilyl)-trifluoroacetamide (MSTFA) | Silylation reagent; replaces active hydrogens with a trimethylsilyl group, conferring volatility [4]. | GC-MS | The most common derivatization agent for GC-MS metabolomics. Often used with catalysts like TMCS. |
| Isotopically Labeled Internal Standards (¹³C, ¹⁵N, ²H-labeled metabolites) | Correct for losses during sample prep and variability in instrument response; enable absolute quantification [1]. | Both (Essential) | Should be added as early as possible in the protocol. Ideally, one standard per metabolite class. |
| Formic Acid / Ammonium Acetate | LC-MS mobile phase additives; improve protonation/deprotonation and ionization efficiency in ESI [25] [1]. | LC-MS | Concentration (typically 0.1%) and pH are critical for optimal chromatographic peak shape and sensitivity. |
| Methyl tert-butyl ether (MTBE) | Organic solvent for liquid-liquid extraction; particularly effective for lipidome extraction [1]. | LC-MS (Lipidomics) | Used in biphasic systems with methanol/water for partitioning lipids from polar metabolites. |
| Solid Phase Microextraction (SPME) Fiber | For headspace sampling of volatile organic compounds (VOCs); adsorbs analytes for thermal desorption into GC [4]. | GC-MS (Volatiles) | Enables sensitive analysis of volatiles without solvent, used in breath, plant, and microbiome research. |
Integrated Workflow and Strategic Application
The path from a biological question to metabolic insight involves a series of critical decisions. The following diagram maps the strategic workflow for a metabolomics study, highlighting key decision points where the choice between GC-MS and LC-MS is paramount.
Decision Framework and Concluding Synthesis
Selecting the optimal platform requires a systematic evaluation of the research objectives.
Table 4: Decision Framework for Platform Selection in Natural Product Metabolomics
| Primary Consideration | Choose GC-MS When: | Choose LC-MS When: | Consider a Complementary Approach When: |
|---|---|---|---|
| Analyte Properties | Targets are volatile, semi-volatile, or can be made volatile via derivatization (e.g., essential oils, short-chain fatty acids, sugars) [4] [15]. | Targets are polar, ionic, thermally labile, or have high molecular weight (e.g., most flavonoids, glycosides, peptides, complex lipids) [15] [14]. | The study aims for comprehensive coverage of a broad metabolome (e.g., plant extract, microbial supernatant). Use both platforms for orthogonal analysis [15]. |
| Identification Priority | High-confidence identification using extensive, standardized spectral libraries is required [4]. | Discovery of unknowns is key, and resources exist for MS/MS interpretation and/or authentic standard purchase [15]. | The sample is limited and precious; pilot studies with both platforms determine the richest source of biomarkers. |
| Quantitative Rigor | Excellent chromatographic resolution and reproducibility are needed for complex mixtures or isomers [15]. | Ultimate sensitivity (low LOD) is required for trace-level biomarkers in blood or tissue [14]. | The project involves both high-sensitivity quantification of specific targets (LC-MS/MS) and profiling of volatile cohorts (GC-MS). |
| Operational Logistics | Budget for instrumentation and consumables is a major constraint [15]. | Sample throughput for complex biological matrices is a priority, and expertise in LC method development is available. | The research group is establishing long-term capabilities; investment in a dual-platform lab maximizes future project flexibility. |
Conclusion: Neither GC-MS nor LC-MS is universally superior; they are orthogonal and complementary techniques. GC-MS remains the "gold standard" for the robust, library-supported analysis of volatile and derivatizable metabolites, offering exceptional chromatographic resolution [4] [15]. LC-MS provides unparalleled breadth in analyzing thermally labile and high-molecular-weight compounds, which constitute a large portion of the natural product space, with superior sensitivity in many targeted bioanalyses [15] [14]. The most powerful metabolomics strategy for comprehensive natural product research often involves deploying both platforms in parallel, thereby leveraging the unique strengths of each to illuminate a more complete picture of the metabolic state.
The metabolome of natural products—encompassing plant extracts, marine organisms, and microbial cultures—represents one of the most chemically diverse spaces in nature. This diversity, featuring molecules ranging from volatile terpenes and non-polar lipids to polar glycosides and thermally unstable alkaloids, presents a fundamental analytical challenge [20] [1]. No single analytical platform can capture this full spectrum, creating a compelling scientific and practical need for multiplatform approaches.
This guide objectively compares the two cornerstone technologies in this endeavor: Gas Chromatography-Mass Spectrometry (GC-MS) and Liquid Chromatography-Mass Spectrometry (LC-MS). Framed within the broader thesis of their complementary roles in natural product metabolomics, we provide a detailed performance comparison, supporting experimental data, and standardized protocols to inform researchers, scientists, and drug development professionals in their methodological selections [28] [20].
Core Analytical Platforms: A Head-to-Head Comparison
GC-MS and LC-MS are built on distinct separation and ionization principles, which directly define their applicability to different chemical classes within the natural product metabolome [20] [21].
- GC-MS relies on the volatilization of analytes. It excels for thermally stable, volatile, or derivatizable compounds (e.g., mono- and sesquiterpenes, certain fatty acids, alkaloids, and sugars after derivatization). Its strengths are high chromatographic resolution, excellent reproducibility, and robust, well-established spectral libraries for compound identification [20] [29].
- LC-MS separates compounds in a liquid phase, making it ideal for non-volatile, thermally labile, and high molecular weight compounds (e.g., polyphenols, glycosides, peptides, and polar lipids). Its primary advantage is broad applicability without the need for derivatization, coupled with high sensitivity for trace analysis [30] [2].
The following table summarizes their core analytical characteristics.
Table 1: Comparison of GC-MS and LC-MS for Natural Product Metabolomics
| Feature | GC-MS | LC-MS | Implication for Natural Product Research |
|---|---|---|---|
| Separation Principle | Volatility & gas-phase interaction | Polarity, size, & liquid-phase interaction | GC-MS is limited to volatile/derivatizable compounds; LC-MS has broader scope [20] [21]. |
| Ionization Method | Electron Impact (EI), Chemical Ionization (CI) | Electrospray Ionization (ESI), Atmospheric Pressure CI (APCI) | EI provides reproducible, library-searchable spectra. ESI/APCI better handle fragile molecules [20] [2]. |
| Ideal Compound Classes | Volatiles, fatty acids, sugars, alcohols (often after derivatization) | Polar compounds, glycosides, polyphenols, high-MW lipids, thermolabile alkaloids | Complementary coverage is required for holistic metabolome profiling [28] [1]. |
| Sample Preparation | Often requires derivatization (e.g., silylation, methoximation) | Typically simpler; protein precipitation, filtration | GC-MS prep is more time-consuming and can introduce artifacts [1] [21]. |
| Spectral Libraries | Extensive, reproducible EI libraries (e.g., NIST) | Less universal; libraries are instrument-/condition-dependent | GC-MS offers more confident identifications for known volatiles [20]. |
| Throughput & Robustness | High throughput; very robust and reproducible | High throughput; robustness can be affected by matrix effects | Both suit large-scale screening; GC-MS may have edge in long-term reproducibility [8]. |
Performance Comparison: Supporting Data from Key Studies
Empirical data underscores the complementary nature of these platforms. The following tables present quantitative results from two representative studies: a clinical metabolomics investigation and a food authentication application.
Table 2: Complementary Metabolite Detection in a Lupus Nephritis Study [28] This study analyzed serum from 50 patients and 50 controls using both platforms.
| Analysis Platform | Total Metabolites Detected | Key Classes Identified | Notes on Coverage |
|---|---|---|---|
| GC-MS | A significant subset of 41 potential biomarkers | Amino acids, organic acids, fatty acids, sugars | Excellent for profiling primary metabolism intermediates. |
| LC-MS | A distinct, significant subset of 41 potential biomarkers | Lipids, peptides, bile acids, sterols | Crucial for detecting higher molecular weight and less volatile species. |
| Combined Approach | 41 differentiated metabolites for pathway analysis | Comprehensive coverage across chemical space | The integrated data provided a more complete pathophysiological picture of the disease. |
Table 3: Platform Application in Geographic Authentication of Garlic [29] This study used GC-MS to distinguish garlic from Brazil and China based on metabolomic profiles.
| Metric | GC-MS Performance | Key Discriminatory Metabolites Identified |
|---|---|---|
| Features Detected | 334 total features; 173 annotated | Demonstrates capability for complex plant profiling. |
| Differentiating Power | 84 features significantly different between origins | Successful classification using chemometrics (PCA, PLS-DA). |
| Biomarkers | Brazilian garlic: Higher in organosulfur compounds (allyl propyl sulfide), certain amino acids (L-arginine).Chinese garlic: Higher in sugars (D-fructose), essential amino acids (L-tryptophan). | Platform was ideal for key volatile and derivatizable markers of origin, flavor, and bioactivity. |
Detailed Experimental Protocols
A reliable multiplatform strategy depends on standardized, optimized workflows for each technology. The following protocols are synthesized from established methodologies [28] [1].
Protocol for GC-MS-Based Untargeted Metabolomics
Sample Preparation (Derivatization is Critical):
- Extraction: Homogenize sample (e.g., plant tissue) in a mixture of methanol, chloroform, and water (e.g., 2.5:1:1 ratio). Vortex and centrifuge to separate phases [1].
- Derivatization (Two-Step):
- Methoximation: Dry an aliquot of the extract. Add 20 μL of methoxyamine hydrochloride in pyridine (15 mg/mL). Incubate at 37°C for 90 minutes with shaking to protect carbonyl groups.
- Silylation: Add 80 μL of N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) with 1% TMCS. Incubate at 70°C for 60 minutes to derivatize hydroxyl, amine, and carboxylic acid groups [28] [29].
- QC Sample: Create a pooled Quality Control (QC) sample from aliquots of all experimental samples. This is run repeatedly throughout the sequence to monitor and correct for instrumental drift [8].
Instrumental Analysis:
- GC: Use a non-polar or low-polarity capillary column (e.g., 5%-phenyl dimethylpolysiloxane). Helium carrier gas. Apply a temperature gradient (e.g., 60°C to 325°C) [29].
- MS: Operate in electron impact (EI) mode at 70 eV. Scan across a relevant m/z range (e.g., 50-600). Use the QC data with algorithms (e.g., Random Forest regression) for post-acquisition correction of long-term signal drift [8].
Protocol for LC-MS-Based Untargeted Metabolomics
Sample Preparation (Designed for Broad Polarity):
- Protein Precipitation/Extraction: Add cold methanol or a methanol/acetonitrile mixture (e.g., 2:1) to a liquid sample (e.g., serum) or tissue extract. Vortex vigorously, incubate at -20°C for 1 hour, and centrifuge at high speed (e.g., 13,000 rpm, 4°C, 15 min) [28].
- Clean-up: Transfer the supernatant, potentially filter through a 0.22 μm membrane, and place in an LC vial.
- QC Sample: As with GC-MS, include a pooled QC sample at regular intervals in the run sequence.
Instrumental Analysis:
- LC: Employ either:
- MS: Use a high-resolution mass spectrometer (e.g., Q-TOF, Orbitrap) [2]. Operate in data-dependent acquisition (DDA) or data-independent acquisition (DIA) mode, switching between full MS and MS/MS scans. Electrospray Ionization (ESI) in both positive and negative modes is standard [9].
Data Processing & Analysis Workflow
Post-acquisition, both platforms share a common bioinformatics pipeline:
- Pre-processing: Use software (XCMS, MZmine, MS-DIAL, or MetaboAnalystR 4.0) for peak picking, alignment, and integration [9].
- Compound Identification: For GC-MS, match EI spectra against commercial libraries (NIST). For LC-MS, use accurate mass, MS/MS fragmentation, and retention time against public (e.g., GNPS, HMDB) or in-house libraries [9] [20].
- Statistical Analysis: Perform multivariate analysis (PCA, PLS-DA) to identify group differences and biomarkers [28] [29].
- Pathway Analysis: Map identified metabolites to biochemical pathways using databases like KEGG [28].
Visualizing the Multiplatform Workflow
Diagram 1: Integrated GC-MS/LC-MS Metabolomics Workflow
Title: Parallel metabolomics workflow for GC-MS and LC-MS analysis.
Diagram 2: Data Processing & Analysis Pipeline
Title: Data analysis pipeline from raw spectra to biological insight.
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 4: Key Reagent Solutions for Multiplatform Metabolomics
| Item | Function | Application Notes |
|---|---|---|
| Methanol (MeOH), Acetonitrile (ACN), Chloroform (CHCl₃) | Organic solvents for metabolite extraction via liquid-liquid partitioning [1]. | Cold mixtures (e.g., MeOH:ACN, 2:1) effectively quench enzymes and precipitate proteins. Ratios are adjusted for polarity coverage. |
| Internal Standards (IS) | Compounds spiked into samples to correct for variability in extraction and analysis [1]. | Use stable isotope-labeled analogs (e.g., ¹³C, ²H) of target metabolites or chemical analogs not found natively (e.g., L-2-chlorophenylalanine). |
| Derivatization Reagents:• Methoxyamine HCl• MSTFA + 1% TMCS | For GC-MS: Increase volatility and thermal stability of polar metabolites [28] [29]. | Methoxyamine protects carbonyls; MSTFA replaces active hydrogens with trimethylsilyl groups. Must be performed under anhydrous conditions. |
| Pooled Quality Control (QC) Sample | A homogenized mixture of all experimental samples, run repeatedly [28] [8]. | Monitors instrument stability. Critical for detecting and correcting signal drift over long sequences using computational tools [8]. |
| Alkane Standard Mix (C7-C30) | Provides reference points for retention time indexing in GC-MS, aiding in compound identification [29]. | Run at the beginning or end of a sequence to calibrate retention times across all samples. |
The evidence confirms that the structural and chemical diversity of natural products necessitates a strategic, complementary analytical approach. LC-MS is the broader, more flexible tool, essential for profiling the vast space of polar and labile secondary metabolites. GC-MS is a highly specialized and robust tool, offering unparalleled resolution and confidence in identification for volatiles and primary metabolites.
The choice between or combination of these platforms should be driven by the specific research question:
- Use GC-MS for studies focused on essential oils, volatiles, primary metabolism, or when library-matching confidence is paramount.
- Use LC-MS for discovery-driven research on complex plant extracts, polar bioactive compounds, or high-molecular-weight metabolites.
- Employ an integrated GC-MS/LC-MS strategy when a comprehensive, systems-level view of the metabolome is required, such as in functional genomics, biomarker discovery, or holistic quality assessment of natural products.
By leveraging their complementary strengths within a rigorous, QC-driven workflow, researchers can fully harness the chemical information encoded in the natural product metabolome, accelerating discovery in pharmacology, food science, and systems biology.
Workflows in Action: From Sample to Data for Natural Products
In the field of natural product metabolomics, the analytical power of Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS) is fundamentally constrained by the initial handling of samples. The metabolome is dynamic; metabolic reactions can continue ex vivo, leading to significant alterations in metabolite profiles if not arrested promptly [31]. Therefore, the protocols for sample collection, quenching, and storage are not mere preliminaries but are critical determinants of data accuracy and biological relevance.
This guide objectively compares these foundational steps as applied within LC-MS and GC-MS workflows. The choice between these platforms often dictates specific preparatory requirements: GC-MS is ideal for volatile, non-polar, or derivatized compounds, while LC-MS covers a broader range of semi-polar to polar, thermally labile substances [14] [32]. Framed within the broader thesis of LC-MS versus GC-MS for metabolomics, we evaluate how each platform's technical demands shape the initial sample treatment to preserve the integrity of different chemical classes within natural matrices [4].
Comparative Analysis: Platform Requirements and Impacts
The selection of an analytical platform directly influences sample preparation strategy. The table below summarizes the core differences between LC-MS and GC-MS that dictate initial sample handling protocols.
Table 1: Comparative Overview of LC-MS and GC-MS in Metabolomics Influencing Sample Preparation
| Aspect | GC-MS Platform | LC-MS Platform | Impact on Sample Collection & Storage |
|---|---|---|---|
| Primary Analyte Suitability | Volatile, thermally stable compounds, or compounds made volatile via derivatization (e.g., organic acids, sugars, certain amino acids) [14] [4]. | Broad range, especially semi-polar to polar, high molecular weight, and thermally labile compounds (e.g., lipids, flavonoids, most pharmaceuticals) [14] [33]. | Dictates the quenching method to preserve target metabolite stability (thermal vs. chemical). |
| Typical Sensitivity | High (down to ∼10⁻¹² mol) [14]. | Very High (down to ∼10⁻¹⁵ mol) [14]. | Demands stringent protocols to minimize contamination and analyte loss during handling, which is more critical for ultra-trace analysis in LC-MS. |
| Key Sample Prep Step | Chemical Derivatization (e.g., trimethylsilylation) is often mandatory to increase volatility and thermal stability [4]. | Derivatization is rarely used. Focus is on efficient extraction and cleanup to reduce matrix effects [31] [33]. | Storage conditions must preserve samples in a state suitable for the subsequent derivatization reaction (e.g., preventing hydrolysis). |
| Critical Pre-Analytical Concern | Loss of volatile analytes during concentration/evaporation steps [4]. | Enzymatic degradation and continued metabolism post-sampling [31]. | Quenching must be immediate and effective, particularly for LC-MS studies of labile pathways. |
| Extraction Solvent Compatibility | Final extract must be compatible with derivatization chemistry and GC injection (typically in non-polar solvents) [4]. | Final extract must be compatible with LC mobile phase and ionization (avoiding non-volatile salts, surfactants) [31] [34]. | Initial storage and quenching solvents must be chosen with the final analytical solvent system in mind. |
Experimental Protocols for Natural Matrices
Universal Foundational Steps
For all natural matrices (tissue, biofluids, cell cultures), consistent initial handling is paramount.
- Collection: Use standardized, clean tools. For tissues, rapid excision and washing in cold saline to remove blood is recommended [34].
- Quenching: The goal is to instantly halt enzymatic activity.
- Storage: Maintain a continuous cold chain. Long-term storage should be at -80°C or in liquid nitrogen. Avoid repeated freeze-thaw cycles by aliquoting samples [31] [34].
Platform-Specific Extraction & Preparation
For GC-MS Analysis:
- Extraction: Use a solvent system that balances polar and non-polar metabolite recovery. A ternary mixture of water, acetonitrile, and isopropanol is effective [4].
- Clean-up: A lipid removal step (e.g., hexane wash) is often crucial post-extraction to prevent accumulation in the GC inlet and interference with derivatization [4].
- Derivatization: The critical step. A common method involves:
- Drying the extract under a gentle stream of nitrogen.
- Adding a methoxyamine solution in pyridine to protect carbonyl groups (oximation).
- Subsequently adding a silylating agent like N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA) to replace active hydrogens with trimethylsilyl groups, rendering metabolites volatile [4] [34].
For LC-MS Analysis:
- Extraction: Optimized for broad metabolite coverage and compatibility with LC.
- A common approach is bi-phasic extraction using methanol/chloroform/water (e.g., Bligh & Dyer method) for comprehensive metabolomics, or mono-phasic methanol/water for polar metabolites [31] [34].
- Protein precipitation with organic solvents like acetonitrile or methanol is standard for biofluids [34].
- Clean-up & Concentration: Solid-phase extraction (SPE) can enrich analytes and remove salts. Extracts are often concentrated via gentle nitrogen blowdown evaporation to prevent degradation of labile compounds [34].
- Reconstitution: The dry extract is reconstituted in a solvent compatible with the initial LC mobile phase (often high aqueous content) [31].
Workflow Visualization: From Sample to Data
The following diagrams map the critical decision points and procedural pathways for sample preparation in LC-MS and GC-MS metabolomics.
Diagram 1: Platform Selection & Sample Prep Workflow. This decision tree illustrates the primary branching point based on metabolite physicochemical properties, leading to distinct quenching and preparation protocols for GC-MS and LC-MS analysis [14] [31] [4].
Diagram 2: Integrated Metabolomics Workflow. For comprehensive metabolome coverage, a single sample aliquot can be split after quenching for parallel GC-MS and LC-MS analysis, with data integrated post-processing [32].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 2: Key Reagents and Materials for Sample Preparation
| Item | Primary Function | Platform Specificity & Notes |
|---|---|---|
| Liquid Nitrogen | Rapid quenching and flash-freezing of tissues and cell pellets to instantly halt metabolism [31] [34]. | Universal. Essential for preserving labile metabolites for both platforms. |
| Cold Methanol (-40°C to -80°C) | Chemical quenching and extraction solvent for cell cultures; precipitates proteins [31]. | LC-MS leaning. Often used in protocols for polar metabolite extraction. |
| Methanol, Chloroform, Water | Components for bi-phasic extraction (e.g., Bligh & Dyer method), separating polar (aqueous) and non-polar (organic) metabolites [34]. | LC-MS. Enables broad, untargeted metabolite coverage. |
| Acetonitrile | Organic solvent for protein precipitation from biofluids (e.g., serum, plasma) [34]. | LC-MS. Effective precipitant with low background in MS. |
| Methoxyamine Hydrochloride | Derivatization reagent for oximation; protects carbonyl groups (ketones, aldehydes) prior to silylation [4]. | GC-MS specific. Critical first step in standard derivatization protocols. |
| BSTFA (with 1% TMCS) | Silylation reagent; replaces active hydrogens with trimethylsilyl groups, conferring volatility for GC analysis [4] [34]. | GC-MS specific. The most common derivatizing agent for metabolomics. |
| Solid-Phase Extraction (SPE) Cartridges | Clean-up and fractionation; remove salts, lipids, or other interferences; can enrich low-abundance metabolites [31] [34]. | Both, but more common in LC-MS. Used for sample cleanup prior to injection. |
| Inert Gas (N₂) Blowdown Evaporator | Gently removes extraction solvents under a stream of inert gas to concentrate samples and prevent oxidation [34]. | Both, critical. Preserves sensitive compounds during concentration; avoids heat degradation. |
| Deuterated Solvents & Internal Standards | Used for NMR analysis and as internal standards in MS for quantification, correcting for extraction and ionization variability [31]. | Both (Quantification). Essential for accurate quantitative results in both platforms. |
The foundational step in natural product metabolomics is the efficient extraction of bioactive compounds from complex biological matrices. This process directly determines the scope and quality of data generated by subsequent analytical platforms, primarily Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS) [20]. The core principle governing extraction is "like dissolves like," where solvent polarity is matched to target metabolite polarity [35] [36]. Polar solvents (e.g., water, methanol) are optimal for extracting alkaloids, flavonoids, and glycosides, while non-polar solvents (e.g., hexane, chloroform) target lipids, sterols, and essential oils [37] [36]. The choice between LC-MS and GC-MS for downstream analysis critically influences the initial extraction and sample preparation strategy. LC-MS excels at analyzing a broad range of non-volatile and thermally labile polar to mid-polar compounds, often requiring less extensive derivatization. In contrast, GC-MS offers high resolution for volatile and thermally stable compounds, often necessitating derivatization steps to increase the volatility of polar metabolites [20] [38] [28]. Thus, tailoring the solvent system is the first decisive action in a workflow aimed at generating comprehensive, platform-specific metabolomic profiles for drug discovery and development.
Comparative Performance of Extraction Methods
Selecting an appropriate extraction technique is crucial for yield, selectivity, and compatibility with LC-MS or GC-MS analysis. The following table compares conventional and modern methods [35] [37] [36].
Table 1: Comparison of Extraction Methods for Natural Products
| Method | Mechanism | Optimal Solvent Polarity | Key Advantages | Key Limitations | Suitability for LC-MS/GC-MS |
|---|---|---|---|---|---|
| Maceration | Passive soaking at room temperature [35]. | Dependent on solvent [35]. | Simple; good for thermolabile compounds [35]. | Time-consuming; low efficiency [35]. | LC-MS: Good for broad profiles. GC-MS: May require concentration. |
| Soxhlet Extraction | Continuous cycling of warm solvent [35] [37]. | Non-polar to mid-polar organic [37]. | High efficiency; established SOPs [37]. | Large solvent volume; long time; high temperature [35] [37]. | GC-MS: Excellent for lipidomics. LC-MS: May need solvent exchange. |
| Ultrasound-Assisted (UAE) | Cavitation disrupts cell walls [35] [37]. | Dependent on solvent [35]. | Faster; improved yield; energy-efficient [37]. | Possible degradation; scale-up challenges [37]. | LC-MS: Excellent for phenolics, antioxidants. |
| Microwave-Assisted (MAE) | Microwave energy heats solvent/sample rapidly [35] [37]. | Dependent on solvent (often polar) [36]. | Very fast; reduced solvent use [35] [36]. | Capital cost; limited penetration depth [36]. | LC-MS: Excellent for fast extraction of polar targets. |
| Supercritical Fluid (SFE) | Uses supercritical CO₂ as tunable solvent [35] [37]. | Non-polar to moderate polar (with modifier) [35]. | Green, clean, selective; easy solvent removal [37] [36]. | High equipment cost; high pressure [37] [36]. | GC-MS: Ideal for essential oils, lipids. LC-MS: Good with modifiers. |
| Pressurized Liquid (PLE) | High temp/pressure to enhance solubility/diffusion [35]. | Dependent on solvent [35]. | Fast; automated; small solvent volume [35]. | High equipment cost [36]. | LC-MS/GC-MS: Excellent for high-throughput automation. |
Tailoring Solvent Systems to Metabolite Polarity
The solvent's chemical nature is the primary determinant of extraction selectivity. The table below guides solvent selection based on target compound polarity and analytical platform.
Table 2: Solvent Selection Guide for Polar and Non-Polar Natural Products
| Solvent Category | Examples | Polarity Index | Target Compound Classes | Typical Extraction Method | Primary Analytical Platform |
|---|---|---|---|---|---|
| Polar Protic | Water, Methanol, Ethanol [35] [36]. | High (Water: 10.2) | Alkaloids, flavonoids, glycosides, phenolic acids, sugars [20] [36]. | Maceration, UAE, MAE, Decoction [35]. | LC-MS (especially HILIC/RPLC) [20]. |
| Polar Aprotic | Acetone, Ethyl Acetate [36]. | Medium-High | Broad range, including many polyphenols [36]. | Maceration, Percolation, UAE [35]. | LC-MS (RPLC). |
| Non-Polar | Hexane, Chloroform, Toluene [37] [36]. | Low | Fatty acids, essential oils, sterols, terpenes, waxes [20] [37]. | Soxhlet, SFE (with pure CO₂) [35] [37]. | GC-MS (often direct analysis) [38] [39]. |
| Modern Green Solvents | Supercritical CO₂, Ionic Liquids, Deep Eutectic Solvents [37] [36]. | Tunable (Low to High) | Tailorable; from lipids (CO₂) to polar phenolics (DES) [36]. | SFE, Modified MAE/UAE [37] [36]. | GC-MS (SFE-CO₂), LC-MS (DES). |
Experimental Protocol: A Comparative Metabolomics Workflow
This protocol, based on a comparative study of lupus nephritis patients, details a parallel extraction and analysis method for LC-MS and GC-MS, highlighting platform-specific sample preparation [28].
Title: Parallel Serum Metabolomics Profiling Using LC-MS and GC-MS. Objective: To comprehensively identify differential metabolites in patient serum using complementary LC-MS and GC-MS platforms. Materials: Serum samples, internal standards (L-2-chlorophenylalanine for both, C17:0 for LC-MS), methanol, acetonitrile, methoxyamine hydrochloride, BSTFA (with 1% TMCS), n-Hexane. Protocol:
- Sample Preparation: Thaw serum on ice. For both platforms, add methanol:acetonitrile (2:1) mixture to precipitate proteins, vortex, ultrasonicate in an ice bath, incubate at -20°C, and centrifuge [28].
- LC-MS Specific Preparation: Transfer supernatant, filter through a 0.22 μm organic phase filter, and place in an LC vial [28].
- GC-MS Specific Derivatization: Dry an aliquot of the supernatant under nitrogen or vacuum. Add methoxyamine hydrochloride in pyridine for oximation (37°C, 90 min). Subsequently, add BSTFA for trimethylsilylation (70°C, 60 min) to increase volatility [28].
- Instrumental Analysis:
- Data Processing: Use software (e.g., AMDIS for GC-MS, proprietary for LC-MS) for peak picking, alignment, and deconvolution. Annotate compounds using spectral libraries (e.g., Fiehn for GC-MS, METLIN for LC-MS) [20] [38]. Perform multivariate statistical analysis (PCA, OPLS-DA) to identify significant biomarkers [28]. Key Outcome: The study identified 41 potential biomarkers, demonstrating that the combined use of LC-MS and GC-MS provided a more comprehensive metabolomic coverage than either platform alone [28].
Visualization of Workflows and Platform Comparison
Diagram 1: Natural Product Extraction to MS Analysis Workflow
Diagram 2: LC-MS vs. GC-MS Core Comparison for Metabolomics
The Scientist's Toolkit: Essential Reagents and Materials
Table 3: Key Research Reagents and Materials for Extraction and Metabolomics
| Item | Function/Description | Key Application |
|---|---|---|
| Methanol & Acetonitrile (HPLC Grade) | Polar aprotic solvents for extraction and LC mobile phases; effective protein precipitants [28]. | Universal extraction solvents for polar metabolites; LC-MS analysis [35] [28]. |
| Hexane & Chloroform (HPLC Grade) | Non-polar solvents for extracting lipids, waxes, and essential oils [37] [36]. | Soxhlet or maceration of non-polar compounds; GC-MS sample preparation [39]. |
| Ethanol (Food/Pharma Grade) | Polar protic, relatively low-toxicity solvent for food/pharma applications [35] [36]. | Maceration, percolation, and UAE of polyphenols and alkaloids [35]. |
| Derivatization Reagents (MSTFA/BSTFA) | Trimethylsilylation agents replace active hydrogens with -Si(CH₃)₃ groups [38]. | Critical for GC-MS analysis of polar metabolites (e.g., sugars, organic acids) to increase volatility [38] [28]. |
| Methoxyamine Hydrochloride | Protects carbonyl groups (aldehydes, ketones) by forming methoximes during derivatization [38]. | Prevents cyclization and multiple peak formation in GC-MS analysis of sugars and keto acids [38]. |
| Solid-Phase Extraction (SPE) Cartridges | Contain sorbents (C18, silica, ion-exchange) for clean-up, fractionation, and concentration [37] [39]. | Removing interferences, desalting, and pre-concentrating samples before LC-MS/GC-MS [39]. |
| Internal Standards (IS) | Deuterated or otherwise non-naturally occurring analogs of target compounds (e.g., d₂₇-myristic acid) [38]. | Correcting for analyte loss during preparation and instrumental variability in quantitative MS [8] [28]. |
In the field of natural product metabolomics, the choice between Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS) is foundational, defining the scope and depth of metabolic profiling. LC-MS has become a predominant platform, celebrated for its broad applicability to diverse, non-volatile, and thermally labile compounds such as lipids, flavonoids, and complex glycosides without the need for extensive sample modification [14] [12]. Its exceptional sensitivity, reaching down to 10⁻¹⁵ mol, allows for the detection of trace-level biomarkers directly from complex biological matrices like plant extracts and biofluids [14] [41].
In contrast, GC-MS is the established technique for analyzing volatile and semi-volatile organic compounds. Its traditional strength lies in the analysis of essential oils, aroma compounds, and environmental volatiles [14] [42]. However, a vast chemical space of polar, thermally stable metabolites—including key classes like amino acids, organic acids, sugars, and certain polyphenols—remains inaccessible to GC-MS in their native state due to low volatility and high polarity [43] [44]. This is where derivatization becomes an imperative chemical step. By masking polar functional groups (e.g., -OH, -COOH, -NH₂), derivatization transforms these non-volatile compounds into volatile, thermally stable analogues suitable for gas-phase separation and analysis [45]. This process effectively extends the powerful capabilities of GC-MS—including superior chromatographic resolution, highly reproducible Electron Impact (EI) fragmentation libraries (e.g., NIST), and robust quantitative performance—deep into the polar metabolome [14] [12] [43]. Consequently, within a comprehensive metabolomics strategy, GC-MS with derivatization is not a competitor to LC-MS but a vital complementary tool. It provides orthogonal data on a distinct yet crucial segment of the metabolome, enabling a more complete systems-level understanding of natural products, from quality control and authentication to the discovery of novel bioactive compounds [41] [43].
Performance and Methodology Comparison: Derivatization GC-MS vs. Alternative Platforms
The decision to employ derivatization GC-MS hinges on a clear understanding of its performance metrics relative to other core metabolomics platforms. The following tables summarize key quantitative and qualitative comparisons.
Table 1: Quantitative Performance Comparison of Core Metabolomics Platforms [14]
| Platform | Typical Sensitivity (mol) | Key Analytical Strengths | Key Limitations for Natural Product Analysis |
|---|---|---|---|
| GC-MS (with derivatization) | 10⁻¹² | High sensitivity; Excellent chromatographic resolution; Universal, reproducible EI spectral libraries (NIST); Robust quantification. | Requires derivatization (extra step, risk of artifacts); Limited to thermally stable, derivatizable compounds; May miss very large or labile metabolites. |
| LC-MS (e.g., RPLC/HILIC) | 10⁻¹⁵ | Exceptional sensitivity; Broad coverage of non-volatile and labile compounds (lipids, glycosides); Minimal sample preparation. | Highly dependent on compound-specific ionization efficiency; Complex spectra; Less universal spectral libraries; Can struggle with very polar small molecules. |
| NMR | 10⁻⁶ | Non-destructive; Provides definitive structural information; Quantitative; Excellent for isotopomer analysis. | Lower sensitivity; Limited dynamic range; Complex mixture deconvolution can be challenging. |
Table 2: Method Characteristics of Derivatization GC-MS vs. LC-MS for Polar Metabolites [14] [46] [12]
| Characteristic | Derivatization GC-MS | LC-MS (for polar metabolites) |
|---|---|---|
| Sample Preparation | Complex: Requires extraction, chemical derivatization (often with drying and redissolution steps). | Simpler: Often requires only extraction and filtration/dilution. |
| Chromatographic Separation | Based on volatility and compound-stationary phase interaction in gas phase. Exceptional peak capacity, especially with GC×GC [47]. | Based on polarity/hydrophobicity (RPLC) or hydrophilic interaction (HILIC) in liquid phase. Very flexible. |
| Ionization | Electron Impact (EI) - Hard ionization, extensive fragmentation, library-matchable spectra. | Electrospray Ionization (ESI) - Soft ionization, primarily generates molecular ions ([M+H]⁺/[M-H]⁻). |
| Detection | Mass spectrometry with universal spectral libraries (NIST). High inter-lab reproducibility. | Mass spectrometry. Tandem MS (MS/MS) crucial for identification. Library matching less straightforward. |
| Ideal Compound Classes | Amino acids, organic acids, sugars, sugar alcohols, fatty acids, certain phenols [43]. | Peptides, lipids, glycosides, alkaloids, most secondary metabolites. |
| Throughput | Derivatization is time-limiting (30-120 min). Analysis runtime can be long for complex mixes. | High potential for throughput; fast LC gradients possible. |
| Data Output | Quantitative concentration data for targeted panels of primary metabolites. Semi-quantitative for untargeted. | Quantitative and qualitative for a vast, untargeted range of metabolites. |
Experimental Protocols: Core Derivatization Workflows for GC-MS Metabolomics
The efficacy of a GC-MS metabolomics study is fundamentally determined by the derivatization protocol. Below are detailed methodologies for two of the most common and reliable approaches used in the analysis of polar metabolites from natural products.
Two-Step Methoxyamination and Silylation for Polar Metabolites
This is the benchmark protocol for comprehensive profiling of primary metabolites (sugars, organic acids, amino acids) [43] [45].
- Objective: To derivative carbonyl groups (in sugars, etc.) via methoxyamination and subsequently replace active hydrogens in hydroxyl, carboxyl, and amine groups with trimethylsilyl (TMS) groups.
- Materials: Dry sample extract, methoxyamine hydrochloride in pyridine, N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) with 1% trimethylchlorosilane (TMCS), alkane standard mixture for Retention Index (RI) calibration.
- Procedure:
- Sample Preparation: Lyophilize or completely dry under nitrogen an aliquot of polar extract (e.g., from 80% methanol/water plant extract) in a glass vial.
- Methoxyamination: Add 50 µL of methoxyamine hydrochloride solution (20 mg/mL in anhydrous pyridine) to the dried residue. Vortex vigorously and incubate at 30°C for 90 minutes with shaking. This step converts aldehydes and ketones to methoximes, preventing multiple isomer formation and improving chromatography.
- Silylation: Add 100 µL of MSTFA (+1% TMCS as catalyst) to the reaction mixture. Vortex and incubate at 37°C for 30 minutes. TMCS catalyzes the complete silylation of all active H atoms.
- Completion & Injection: The derivatized sample is stable for 24-48 hours at room temperature. Transfer an aliquot to a GC vial with insert for analysis.
- GC-MS Parameters: Inlet: 250°C, split or splitless mode; Column: Mid-polarity stationary phase (e.g., 35% phenyl / 65% dimethylpolysiloxane); Oven: Ramp from 60°C to 330°C; Carrier: Helium; MS: EI at 70 eV, scan range m/z 50-600.
The following diagram illustrates the logical workflow and critical decision points in this protocol.
Workflow for Two-Step Methoxyamination and Silylation
Methyl Chloroformate (MCF) Derivatization for Amino Acids and Organic Acids
This protocol is favored for its rapid, room-temperature reaction and specific application to amino and organic acids, often in physiological samples [46].
- Objective: To derivative carboxylic acid and amine groups simultaneously, forming volatile methyl ester and carbamate derivatives.
- Materials: Aqueous sample (e.g., plant sap, urine), sodium hydroxide, methanol, pyridine, methyl chloroformate (MCF).
- Procedure:
- Alkalization & Alcoholysis: Mix 50 µL of sample with 100 µL of sodium hydroxide (1M) and 167 µL of methanol in a vial. Vortex.
- Derivatization: Add 34 µL of pyridine, followed by slow, dropwise addition of 20 µL of methyl chloroformate under continuous vortexing (CAUTION: exothermic). Cap and vortex for an additional 30 seconds.
- Extraction: Add 400 µL of chloroform to extract the derivatized products, followed by 400 µL of sodium bicarbonate solution (50 mM) to quench the reaction and remove excess reagent. Vortex and centrifuge.
- Analysis: Collect the lower organic (chloroform) layer containing the derivatives for GC-MS analysis.
- Key Advantages & Caveats: This method is fast (<5 min) and occurs in aqueous medium. However, it requires careful pH control; highly alkaline conditions can cause racemization of chiral amino acids, while proteins in the sample can hydrolyze, skewing "free" amino acid measurements [46]. Protein precipitation prior to derivatization is recommended for accurate free amino acid profiling.
The Scientist's Toolkit: Key Reagents for Derivatization
Successful derivatization depends on selecting the appropriate reagent for the target functional groups and analytical goals. The table below details the most critical reagents.
Table 3: Key Derivatization Reagents for GC-MS Metabolomics [46] [44] [45]
| Reagent Category | Specific Reagents | Target Functional Groups | Key Function & Outcome |
|---|---|---|---|
| Silylation Agents | MSTFA (N-Methyl-N-(trimethylsilyl)trifluoroacetamide), BSTFA (N,O-Bis(trimethylsilyl)trifluoroacetamide) | -OH, -COOH, -NH, -SH | Replaces active H with a -Si(CH₃)₃ group. Drastically reduces polarity and boiling point. The most common approach for comprehensive metabolomics [45]. |
| Acylation Agents | PFPA (Pentafluoropropionic anhydride), HFBA (Heptafluorobutyric anhydride) | -NH₂, -OH | Adds a perfluorinated acyl group. Increases volatility and enhances electron-capture detector (ECD) or negative chemical ionization (NCI) sensitivity due to fluorine content [46]. |
| Alkylation Agents | Methyl Chloroformate (MCF), TMAH (Tetramethylammonium hydroxide) | -COOH (to methyl esters), -NH₂ (to carbamates with MCF) | Adds alkyl groups (e.g., CH₃). MCF allows fast, aqueous-phase derivatization. TMAH can perform online thermochemolysis in the GC inlet [46] [45]. |
| Chiral Derivatization Agents | Chiral chloroformates, Chiral alcohols (after activation) | -NH₂, -OH | Reacts with enantiomers to form diastereomers, which can be separated on a standard, non-chiral GC column, enabling enantiomeric ratio analysis [45]. |
Strategic Selection: When to Use Derivatization GC-MS in a Research Pipeline
The choice to implement derivatization GC-MS should be driven by specific research questions and sample properties. The following diagram provides a strategic decision framework for method selection in natural product metabolomics.
Strategic Decision Guide for Analytical Platform Selection
Guiding Principles for Application:
- Choose Derivatization GC-MS when: The research prioritizes absolute quantification of known primary metabolites (e.g., in flux analysis), requires maximum chromatographic resolution for complex mixtures of small polar molecules, or demands high inter-laboratory reproducibility through the use of universal EI libraries [47] [43]. It is the method of choice for projects focusing on energy metabolism, osmotic regulation, or quality control based on specific acidic/amino acid profiles [41].
- Choose LC-MS when: The target analytes are inherently non-volatile and thermally labile (e.g., large polyphenols, peptides, most triglycerides), when a broad, untargeted discovery of unknown secondary metabolites is the goal, or when sample throughput must be maximized and derivatization steps avoided [14] [12].
- Employ a Multi-Platform Strategy when: A comprehensive, systems-level view of the metabolome is essential. For instance, using derivatization GC-MS to precisely quantify central carbon metabolites while employing LC-MS to profile lipids and secondary signaling molecules provides unparalleled coverage and is considered a gold-standard approach in advanced natural product and functional metabolomics studies [41] [12].
In natural product metabolomics research, the choice of analytical platform fundamentally shapes the scope and reliability of the metabolic profile obtained. The core of this decision lies in the separation technique—gas chromatography (GC) or liquid chromatography (LC)—coupled to mass spectrometry (MS), and more specifically, in the ionization method that bridges the separation step to mass detection [48] [21]. Electron Ionization (EI), used in GC-MS, and Electrospray Ionization (ESI) or Atmospheric Pressure Chemical Ionization (APCI), used in LC-MS, operate on fundamentally different physical and chemical principles. These differences dictate the classes of metabolites that can be analyzed, the type of spectral information generated, and the overall workflow, from sample preparation to data interpretation [21] [49].
This guide provides a detailed, objective comparison of these ionization techniques. It is framed within the critical thesis of selecting between LC-MS and GC-MS platforms for metabolomics, a field that demands comprehensive coverage of chemically diverse small molecules to understand biological systems [48]. We compare performance parameters such as analyte coverage, sensitivity, spectral reproducibility, and susceptibility to matrix effects, supported by experimental data from recent studies. The goal is to equip researchers and drug development professionals with the knowledge to select the optimal ionization strategy for their specific natural product research questions.
Fundamental Principles and Mechanisms
The ionization process is the critical step that converts neutral analyte molecules into gas-phase ions suitable for mass analysis. The mechanism profoundly influences the technique's applicability.
Electron Ionization (EI) in GC-MS: EI is a hard, high-vacuum ionization method. Analytes, eluted from the GC column into the ion source, are bombarded with a beam of 70 eV electrons. This high-energy interaction typically removes an electron from the analyte molecule, generating a radical cation (M⁺•). The excess internal energy often causes extensive and reproducible fragmentation [49]. While this fragmentation can obscure the molecular ion, it provides rich structural "fingerprint" spectra that are highly consistent across instruments, enabling powerful library-based identification [49]. A key limitation is that EI requires analytes to be in the gas phase, restricting analysis to volatile, thermally stable compounds or those that can be made volatile through chemical derivatization [21].
Electrospray Ionization (ESI) in LC-MS: ESI is a soft, atmospheric-pressure ionization technique. The LC eluent is sprayed through a charged needle to form a fine aerosol of charged droplets. As the solvent evaporates, charged analyte molecules (e.g., [M+H]⁺ or [M-H]⁻) are ejected into the gas phase [50] [51]. This process is highly efficient for pre-charged or easily protonated/deprotonated molecules, such as organic acids, alkaloids, and most polar metabolites. However, it is less effective for neutral, nonpolar compounds and is highly susceptible to ion suppression or enhancement from co-eluting matrix components [52] [51].
Atmospheric Pressure Chemical Ionization (APCI) in LC-MS: APCI is also a soft ionization technique operating at atmospheric pressure. The key difference is that the LC eluent is nebulized and vaporized in a heated tube (typically >350°C) to form a gas-phase mist. A corona discharge needle then ionizes the solvent vapor, initiating gas-phase chemical reactions (e.g., proton transfer) that ultimately ionize the analyte molecules [50] [51]. APCI is more effective than ESI for less polar, thermally stable compounds of low to medium molecular weight and generally exhibits reduced matrix effects because ionization occurs in the gas phase after solvent/analyte separation [52] [53].
Diagram 1: Fundamental Ionization Mechanisms for EI, ESI, and APCI [50] [49] [51].
Performance Comparison: Analyte Coverage, Sensitivity, and Selectivity
The complementary nature of these techniques is best illustrated by their performance across different chemical spaces. The following tables consolidate quantitative and qualitative data from comparative studies.
Table 1: Comparative Performance of Ionization Techniques Based on Analyte Properties [54] [50] [52]
| Performance Aspect | GC-MS with EI | LC-MS with ESI | LC-MS with APCI |
|---|---|---|---|
| Optimal Analyte Class | Volatile, thermally stable, non-polar to mid-polar (e.g., hydrocarbons, sterols, pesticides, derivatized sugars/acids). | Polar, ionic, and high molecular weight compounds (e.g., organic acids, flavonoids, glycosides, peptides). | Low to medium polarity, thermally stable compounds (e.g., lipids, less polar aglycones, some sterols, PAHs). |
| Ionization Mechanism | High-energy electron bombardment (70 eV). | Ion evaporation from charged droplets. | Gas-phase chemical ionization after thermal vaporization. |
| Typical Ions Formed | Radical cation (M⁺•), extensive fragments. | Protonated ([M+H]⁺) or deprotonated ([M-H]⁻) molecules, adducts. | Protonated ([M+H]⁺) or deprotonated ([M-H]⁻) molecules. |
| Molecular Ion Information | Often weak or absent; relies on fragments. | Intact molecular ion (or adduct) is dominant. | Intact molecular ion is typically observed. |
| Library Searchability | Excellent. Reproducible spectra enable large database searches (e.g., NIST). | Poor. Spectra are instrument and condition dependent. | Poor. Not suitable for library matching. |
| Susceptibility to Matrix Effects | Generally low. Separation occurs before gas-phase ionization. | High. Co-eluting matrix can suppress/enhance ionization in the droplet. | Moderate. Reduced compared to ESI, as ionization is gas-phase. |
Table 2: Experimental Sensitivity and Selectivity Data from Comparative Studies
| Study Context | Technique | Key Performance Finding | Reference |
|---|---|---|---|
| Pesticides in Air Samples | GC-EI/SIM | Method Detection Limits (MDLs) of 1–10 pg/μL for OPs (e.g., phorate) and OCs (e.g., DDT) with poor NCI response. | [54] |
| GC-NCI/SIM | Lowest MDLs (2.5–10 pg/μL) for most pesticides; best confirmation via ion ratios. | [54] | |
| Grape Metabolomics | LC-ESI-MS | Lower Limits of Detection (LODs) for polar metabolites (sucrose, tartaric acid) but narrower linear range & greater matrix effects vs. APCI. | [50] |
| LC-APCI-MS | More suitable for strongly polar sugars/acids; generated more fragment ions; complementary coverage to ESI. | [50] | |
| Pharmaceutical Analysis (Levonorgestrel) | LC-ESI-MS/MS | Lower LLOQ (0.25 ng/mL) compared to APCI (1 ng/mL), but more susceptible to matrix effects. | [53] |
| LC-APCI-MS/MS | Less liable to matrix effects from human plasma matrix. | [53] | |
| Multiclass Pesticide Screening | LC-FμTP-MS | 70% of pesticides had higher sensitivity vs. ESI; 76-86% showed negligible matrix effects (vs. 35-67% for ESI). | [52] |
Key Insights from Comparative Data:
- No single technique is universal. For example, in pesticide analysis, GC-NCI provided the lowest detection limits for many compounds, but GC-EI was essential for others like phorate and DDT [54]. Similarly, in plant metabolomics, ESI and APCI provided complementary coverage of the metabolome [50].
- Matrix effects are a critical differentiator. ESI is notably prone to ion suppression in complex matrices like food extracts or plasma [52] [53]. APCI and newer plasma-based techniques like FμTP demonstrate significantly reduced matrix effects, enhancing quantitative reliability [52].
- The "chemical space" is expanded by combining techniques. A study on crude oil components concluded that no single source (ESI, APCI, or APPI) could ionize all model compounds, highlighting the need for complementary approaches for comprehensive analysis [51].
Experimental Protocols for Key Comparative Studies
To ensure reproducibility and provide a practical foundation, here are detailed methodologies from pivotal comparative studies.
Objective: To evaluate the performance of ESI and APCI for LC-MS-based metabolomic analysis of Corvina grape berry extracts across ripening stages.
Sample Preparation:
- Collection & Quenching: Grape berries were collected at veraison, early-ripening, and full-ripening stages over two growing seasons.
- Extraction: Frozen berry powder was extracted with methanol. The extract was centrifuged, and the supernatant was filtered prior to LC-MS analysis.
LC-MS Analysis:
- Chromatography: Reversed-phase liquid chromatography (likely C18 column) was employed.
- Ionization Comparison: The same sample set was analyzed in separate runs using ESI and APCI sources on the same instrument.
- Mass Spectrometry: Full-scan data was acquired in both positive and negative ionization modes.
- Data Processing: Features were extracted, aligned, and assembled into a data matrix for multivariate statistical analysis (e.g., PCA).
Performance Evaluation Metrics:
- Untargeted: Multivariate analysis of 608 metabolic features to assess clustering and coverage.
- Targeted: Determination of LODs, LOQs, linear range, and matrix effects for specific metabolites (sucrose, tartaric acid, caftaric acid, etc.).
Objective: To compare detection limits of GC-MS (SIM) and GC-MS/MS (SRM) using both EI and Negative Chemical Ionization (NCI) for trace pesticide analysis in air samples.
Sample Preparation:
- Air Sampling: Air samples were collected using high-volume air samplers equipped with polyurethane foam (PUF) and XAD-2 resin cartridges.
- Extraction: Cartridges were Soxhlet-extracted with acetone followed by petroleum ether. Extracts were concentrated and solvent-exchanged to isooctane.
GC-MS Analysis:
- Chromatography: Analysis was performed using gas chromatography.
- Ionization & Detection Modes: Four methods were compared on the same system:
- GC-EI/SIM: Electron Ionization with Selected Ion Monitoring.
- GC-NCI/SIM: Negative Chemical Ionization with SIM.
- GC-NCI/SRM: NCI with Selected Reaction Monitoring (tandem MS).
- GC-EI/SRM: EI with SRM.
- Performance Measurement: Method Detection Limits (MDLs) were determined for over 50 pesticides (organochlorines, organophosphates, herbicides).
Objective: To evaluate ESI and APCI sources for the LC-MS/MS determination of levonorgestrel in human plasma and select the best method for a pharmacokinetic study.
Sample Preparation:
- Extraction: 500 µL of human plasma underwent liquid-liquid extraction (LLE) with 4 mL cyclohexane after the addition of internal standard (canrenone) and saturated sodium bicarbonate.
- Reconstitution: The organic layer was evaporated to dryness and reconstituted in mobile phase.
LC-MS/MS Analysis:
- Chromatography: A C18 column was used with an isocratic mobile phase of methanol and 0.01% formic acid (80:20, v/v). Flow rates were optimized separately for ESI (0.2 mL/min) and APCI (1.0 mL/min).
- Ionization Comparison: The same sample extracts were analyzed using ESI and APCI sources on a triple quadrupole mass spectrometer.
- Performance Evaluation: Methods were compared based on sensitivity (LLOQ), linearity, and matrix effects (assessed by post-extraction spike method).
Workflow Integration: Selecting an Ionization Strategy
The choice of ionization technique is not isolated; it is intrinsically linked to the initial sample properties, research goals, and the overall analytical workflow. The following decision logic, based on characteristics of natural products, can guide platform selection.
Diagram 2: Decision Workflow for Selecting Ionization Techniques in Natural Product Metabolomics [50] [21] [49].
The Scientist's Toolkit: Essential Research Reagents and Materials
Successful implementation of these ionization techniques relies on specific reagents, solvents, and consumables. The following toolkit is compiled from protocols in the cited studies.
Table 3: Essential Research Reagent Solutions for Ionization Techniques
| Item | Primary Function | Common Use Case & Technique | Key Considerations |
|---|---|---|---|
| Methanol (MeOH), Acetonitrile (ACN), Water (H₂O) | HPLC-grade solvents form the mobile phase for LC separation and are the primary solvents for ESI/APCI. | LC-MS (ESI/APCI): Mobile phase preparation, sample reconstitution, metabolite extraction [50] [53]. | Low volatility for ESI droplet formation; purity is critical to reduce background noise and ion suppression. |
| Formic Acid, Ammonium Acetate, Ammonium Hydroxide | Mobile phase additives. Acids promote positive ionization ([M+H]⁺); bases promote negative ionization ([M-H]⁻). | LC-MS (ESI/APCI): Modifying mobile phase pH to optimize ionization efficiency and chromatographic peak shape [53]. | Concentration is typically 0.1% or low mM; volatile buffers are required to avoid source contamination. |
| Derivatization Reagents(e.g., MSTFA, BSTFA) | Chemically modify polar functional groups (e.g., -OH, -COOH) to increase volatility and thermal stability. | GC-MS (EI): Preparation of non-volatile metabolites (sugars, organic acids) for GC analysis [21]. | Reaction conditions (time, temperature) must be optimized. Reagents must be anhydrous. |
| Helium (He) Gas | Ultra-high-purity carrier gas for GC. Also used as a damping/collision gas in MS. | GC-MS (EI): Mobile phase for gas chromatography [21] [49]. | Purity (>99.999%) is essential for sensitivity and column longevity. Supply instability is a growing concern [52]. |
| Internal Standards (IS)(e.g., stable isotope-labeled analogs) | Added to samples at known concentration to correct for variability in extraction, ionization, and instrument response. | All Techniques (Quantitative): Essential for reliable quantification in complex matrices [48] [53]. | Should be chemically identical to analyte but distinguished by mass (e.g., ¹³C, ²H). Added at the start of sample prep. |
| QuEChERS Kits(MgSO₄, NaCl, PSA, C18, EMR-lipid) | Dispersive solid-phase extraction (dSPE) salts and sorbents for rapid cleanup of complex food/plant matrices. | LC/GC-MS Sample Prep: Removing sugars, fatty acids, pigments, and other interferences that cause matrix effects [52]. | Sorbent choice is matrix-dependent (e.g., EMR-lipid for high-fat samples). |
| Perfluorotributylamine (PFTBA) | Calibration standard for mass spectrometers. Provides characteristic ions across a known mass range. | GC-MS (EI) Tuning: Used to calibrate mass axis and optimize instrument parameters (lens voltages) for sensitivity and resolution [49]. | Standard for autotune procedures. Should be used regularly to ensure instrument performance. |
The comparison between EI for GC-MS and ESI/APCI for LC-MS underscores a central theme in modern natural product metabolomics: technique complementarity, not replacement. EI provides unparalleled structural information and standardized spectral libraries for volatile compounds, while ESI and APCI offer soft ionization for a vast range of polar and semi-polar biomolecules directly from liquid streams [50] [49]. The decision must be driven by the chemical nature of the target metabolome, the required data quality (identification vs. quantification), and the complexity of the sample matrix.
Future directions aim to bridge the gaps highlighted in this comparison. Advanced GC-MS techniques like Cold EI use supersonic molecular beams to analyze less volatile compounds and provide enhanced molecular ions, effectively narrowing the application gap with LC-MS [49] [55]. On the LC-MS front, the development of novel plasma-based ionization sources (e.g., FμTP) promises broader chemical coverage and significantly reduced matrix effects compared to traditional ESI [52]. Furthermore, dual-column LC-MS systems that combine orthogonal separation mechanisms (e.g., reversed-phase and hydrophilic interaction chromatography) are emerging as powerful tools to expand metabolite coverage within a single analytical run [27].
For researchers undertaking a thesis on LC-MS versus GC-MS for natural product metabolomics, the most robust strategy may be a multi-platform approach. A workflow that intelligently combines the strengths of GC-EI for volatile and derivatized metabolites with the versatility of LC-ESI/APCI for polar and high-molecular-weight compounds will deliver the most comprehensive and chemically realistic picture of the metabolome [21]. Ultimately, the selection of ionization and detection technology is the foundational step that determines the breadth, depth, and reliability of metabolic insight.
The quality control of natural product (NP)-derived medicines has historically relied on morphological assessment or the quantification of one or two marker compounds using techniques like HPLC [41]. However, NPs are complex mixtures of hundreds of metabolites, making single-marker approaches insufficient for detecting adulteration or authenticating species [41]. Metabolomics, the comprehensive analysis of small-molecule metabolites, has emerged as a powerful solution, with chromatography-mass spectrometry being the core technological platform [41].
The choice between the two principal techniques—Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS)—is foundational to research design. LC-MS excels at analyzing non-volatile, thermally labile, and high-molecular-weight compounds (e.g., proteins, peptides, and many glycosides) with minimal sample preparation [12] [3]. It is the dominant tool for broad metabolomic surveys, especially in biological fluids [12]. In contrast, GC-MS is the "gold standard" for volatile and semi-volatile compounds, offering superior separation efficiency and access to robust, standardized spectral libraries [4] [56]. For non-volatile metabolites like organic acids, sugars, and fatty acids, GC-MS requires a chemical derivatization step to increase volatility [4]. This requirement, once seen as a drawback, provides a structured analytical window that yields highly reproducible data, making GC-MS uniquely powerful for standardized authentication and profiling.
This guide objectively compares the performance of GC-MS and LC-MS within NP metabolomics, using a landmark study on medicinal seahorse authentication as a focused application spotlight [57].
Performance Comparison: GC-MS vs. LC-MS for Metabolomics
The selection between GC-MS and LC-MS is guided by the chemical nature of the analytes and the specific research goals. The following table summarizes their core technical and performance characteristics.
Table 1: Core Performance Comparison of GC-MS and LC-MS for Natural Product Metabolomics
| Aspect | GC-MS | LC-MS |
|---|---|---|
| Ideal Analyte Profile | Volatile, semi-volatile, or derivatizable compounds (e.g., fatty acids, sugars, organic acids, sterols, essential oils). Thermally stable [3] [56]. | Non-volatile, thermally labile, polar, and high-molecular-weight compounds (e.g., peptides, proteins, glycosides, most polyphenols) [12] [3]. |
| Separation Principle | Gas chromatography. Separation based on volatility and interaction with column stationary phase [12]. | Liquid chromatography (often reverse-phase). Separation based on polarity, hydrophobicity, and affinity [12]. |
| Typical Ionization Source | Electron Ionization (EI) [12]. | Electrospray Ionization (ESI) or Atmospheric Pressure Chemical Ionization (APCI) [12]. |
| Key Strengths | Highly reproducible, fragment-rich EI spectra; Extensive, searchable spectral libraries (e.g., NIST); Excellent chromatographic resolution and precision for quantitative analysis [4] [56]. | Broad analyte coverage without derivatization; Superior sensitivity for trace-level biomolecules; Capability to analyze intact large molecules [12] [3]. |
| Primary Limitations | Requires derivatization for most primary metabolites; Limited to thermally stable, lower MW compounds (<~650 Da) [4]. | Less reproducible fragment spectra; Smaller, less universal spectral libraries; Can be susceptible to ion suppression from complex matrices [12]. |
| Best Suited for Authentication When... | The distinguishing chemical markers are volatile or belong to chemical classes like fatty acids, alcohols, or essential oils. Standardized, library-based identification is required [57] [4]. | Markers are large, polar, or non-derivatizable (e.g., specific saponins, alkaloids, phospholipids). A discovery-oriented, untargeted profile is the goal. |
Application Spotlight: Seahorse Species Authentication
Medicinal seahorse (Hippocampus) is a valuable animal-derived drug in traditional medicine. The market is plagued by substitution, where the genuine Hippocampus kelloggi (HK) is replaced by lower-cost species like Hippocampus ingens (HI), compromising clinical safety and efficacy [57]. Morphological identification is error-prone, creating a need for reliable chemical analysis.
A 2023 study pioneered a GC-MS fingerprinting combined with chemical pattern-recognition strategy to differentiate HK from HI [57]. The experimental workflow and key findings are detailed below.
Table 2: Key Experimental Findings from GC-MS Seahorse Authentication Study [57]
| Analysis Method | Finding | Implication for Authentication |
|---|---|---|
| GC-MS Fingerprinting & Hierarchical Clustering (HCA) | All 34 samples clustered perfectly by species based on characteristic peak profiles. | GC-MS fingerprints provide a global chemical profile sufficient for species differentiation. |
| Unpaired t-test (p < 0.0001) | Significant differences in relative content of 10 fatty acids, including lauric, palmitoleic, EPA, and DHA. | Quantitative differences in common metabolites can serve as secondary markers. |
| Orthogonal Projections to Latent Structures Discriminant Analysis (OPLS-DA) | Identified 7 fatty acids (e.g., DHA, EPA, n-hexadecanoic acid) as major discriminatory variables (VIP >1). | Chemometrics pinpoints the most influential chemical markers for building a robust classification model. |
| Targeted Comparison | Nonadecanoic acid and behenic acid were detected exclusively in HK samples. | These compounds are diagnostic chemical markers for the genuine species (HK). |
Experimental Protocol Summary [57]:
- Sample Preparation: Dried seahorse powder was subjected to lipid extraction. A derivatization step using N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA) was critical to make non-volatile fatty acids and other metabolites volatile for GC-MS analysis.
- GC-MS Analysis: Analysis was performed on a system equipped with a non-polar HP-5MS column. Helium was the carrier gas. Electron Ionization (EI) at 70 eV generated characteristic fragment spectra.
- Data Processing & Chemometrics: Total ion chromatograms (fingerprints) were processed. The relative content of characteristic peaks was used as input for multivariate statistical analysis, including HCA, PCA, and OPLS-DA, to identify clustering patterns and key markers.
Diagram 1: GC-MS Seahorse Authentication Workflow
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Research Reagent Solutions for GC-MS Metabolomic Authentication
| Item | Function in Workflow | Example/Chemical Class |
|---|---|---|
| Derivatization Reagent | Converts polar, non-volatile functional groups (-OH, -COOH, -NH₂) into volatile, thermally stable derivatives for GC analysis. | BSTFA (N,O-Bis(trimethylsilyl)trifluoroacetamide) with TMCS catalyst; Methoxyamine hydrochloride [4]. |
| Internal Standard (IS) | Added at sample start to correct for variability in extraction, derivatization, and instrument response; essential for quantification. | Stable isotope-labeled compounds (e.g., ¹³C-sugars, D₄-succinic acid) or non-native analogues (e.g., ribitol for sugars) [4]. |
| Extraction Solvents | To comprehensively extract metabolites of diverse polarities from the complex natural product matrix. | Ternary mixtures (e.g., methanol:acetonitrile:water or isopropanol:acetonitrile:water) are common [4]. |
| GC-MS Spectral Library | Reference database of EI mass spectra and retention indices for compound identification. | NIST Mass Spectral Library, FiehnLib, Golm Metabolome Database [4]. |
| Quality Control (QC) Pool | A pooled sample of all experimental extracts, run repeatedly throughout the sequence. | Monitors instrument stability, validates system suitability, and corrects for signal drift in untargeted analysis [4]. |
Contextualizing the Spotlight: LC-MS vs. GC-MS in a Broader Metabolomics Workflow
The seahorse case exemplifies the targeted application of GC-MS. The broader field of NP metabolomics often employs a complementary, multi-platform strategy. LC-MS is frequently the first choice for untargeted, discovery-phase profiling due to its wide analyte coverage [12] [58]. When hypotheses or specific chemical classes (like fatty acids in seahorse) are identified, GC-MS provides superior standardization and library-based verification [4].
The integration of these platforms is reflected in market and technology trends. The mass spectrometry market is growing robustly, driven by pharmaceutical and biotechnology demand [59] [60]. Key technological trends include the rise of high-resolution and hybrid systems (e.g., Q-TOF, Orbitrap) and the integration of AI for data processing [60], which benefit both LC-MS and GC-MS workflows.
Diagram 2: Strategic Selection between LC-MS and GC-MS
The authentication of medicinal seahorse via GC-MS fingerprinting is a powerful demonstration of targeted metabolomics solving a real-world problem. This application highlights the unique strengths of GC-MS: the generation of reproducible, library-searchable data that yields definitive, chemical marker-based answers.
Within the broader thesis of LC-MS versus GC-MS for NP research, the techniques are complementary. LC-MS is the versatile tool for exploratory, wide-lens discovery, often on complex, polar extracts. GC-MS is the precision instrument for focused analysis, providing unparalleled standardization and reliability for specific compound classes. The choice is not hierarchical but strategic, dictated by the chemical nature of the analytical question. For authentication tasks where markers may be volatile or amenable to derivatization—as in the case of fatty acids for seahorse—GC-MS establishes a compelling, data-driven standard for quality control and species conservation [57].
Analytical Performance: LC-MS vs. GC-MS for Metabolomics
The selection of an analytical platform is foundational to metabolomics research. The following tables compare the core technical capabilities, operational costs, and practical performance of Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS) in the context of natural product and biomarker research.
Table 1: Core Technical Comparison of LC-MS and GC-MS [3] [61] [62]
| Parameter | Liquid Chromatography-Mass Spectrometry (LC-MS) | Gas Chromatography-Mass Spectrometry (GC-MS) |
|---|---|---|
| Chromatographic Principle | Separation in a liquid mobile phase (solvents/buffers) based on polarity, size, and affinity [3] [61]. | Separation in an inert gas mobile phase (e.g., helium) based on volatility and interaction with a heated column [3] [61]. |
| Ideal Analyte Profile | Non-volatile, thermally labile, polar, and high molecular weight compounds (e.g., peptides, lipids, most pharmaceuticals) [3]. | Volatile and semi-volatile, thermally stable, low-to-medium molecular weight compounds [3] [62]. |
| Required Sample Derivatization | Typically not required, enabling analysis of native compounds [3]. | Often mandatory for non-volatile or polar compounds to increase volatility and thermal stability [3]. |
| Primary Ionization Techniques | Electrospray Ionization (ESI), Atmospheric Pressure Chemical Ionization (APCI) [2] [3]. | Electron Ionization (EI), Chemical Ionization (CI) [63]. |
| Key Strengths | Broad analyte coverage without derivatization; gentle on labile compounds; superior for large biomolecules [2] [3]. | Excellent separation efficiency and reproducibility; extensive, searchable spectral libraries; robust and standardized [3] [63]. |
| Key Limitations | Higher operational complexity and cost; can be susceptible to ion suppression in complex matrices [3] [61]. | Limited to volatile/derivatizable compounds; high temperatures may degrade thermolabile analytes [3] [28]. |
Table 2: Operational Cost and Practical Considerations [3] [61]
| Consideration | LC-MS | GC-MS |
|---|---|---|
| Instrument Acquisition Cost | Generally higher, especially for high-resolution tandem systems (e.g., Q-TOF, Orbitrap) [3]. | Generally lower for standard configurations [61]. |
| Operational Cost | Higher due to solvent purchase, disposal, and more frequent maintenance of LC components and ionization sources [3] [61]. | Lower, primarily involving carrier gases and routine column/maintenance [61]. |
| Operator Expertise | Requires more specialized training for method development, troubleshooting, and data interpretation [61]. | Considered easier to operate with more standardized methods [61]. |
| Analysis Time | Can be longer due to column equilibration and gradient elution but highly variable [62]. | Typically faster run times due to gas-phase separation [62]. |
| Sample Throughput | High throughput possible with modern ultra-high-performance LC (UHPLC) and automation [2]. | High throughput for volatile compounds without complex preparation [3]. |
Table 3: Performance Metrics in Comparative Metabolomics Studies
| Metric | LC-MS Performance (Evidence) | GC-MS Performance (Evidence) |
|---|---|---|
| Number of Metabolites Identified | In a multi-platform study of Artemisia argyi, LC-MS (UHPLC-Q-TOF-MS) identified 32 major non-volatile compounds [6]. | In the same study, GC-MS identified 66 volatile compounds [6]. |
| Biomarker Discovery Yield | In a lupus nephritis serum study, LC-MS contributed to a panel of 41 potential biomarkers related to immune regulation and energy metabolism [28]. | GC-MS was used complementarily in the same study to expand metabolome coverage [28]. |
| Publication & Adoption Trend | Higher yearly publication rate (approx. 3908 articles/year from 1997-2023), with a rising LC-MS/GC-MS publication ratio (1.5:1 in 2024) [63]. | Stable, linear publication rate (approx. 3042 articles/year from 1995-2023) [63]. |
| Role in Clinical Diagnostics | Driving growth in precision medicine diagnostics market due to high sensitivity and multiplexing capability for proteins, hormones, and drugs [64]. | Established role in screening for inborn errors of metabolism and quantifying volatile biomarkers [63]. |
Experimental Protocols for Biomarker Discovery
The power of LC-MS in broad profiling is best demonstrated through standardized workflows. The following protocols detail the key phases of discovery and validation, as applied in contemporary research such as acute myeloid leukemia (AML) studies [65].
Protocol 1: Untargeted LC-MS Profiling for Biomarker Discovery
- Objective: To comprehensively and non-selectively profile metabolites/proteins in case vs. control samples (e.g., AML patient vs. healthy serum) [65] [1].
- Sample Preparation:
- Quenching & Extraction: Use rapid cold methanol quenching (e.g., -80°C) to halt enzyme activity. Employ a biphasic liquid-liquid extraction (e.g., methanol/chloroform/water) to broadly capture both polar and non-polar metabolites [1].
- Clean-up: Deplete high-abundance proteins (e.g., albumin) using immunoaffinity columns to enhance detection of low-abundance targets [65].
- Internal Standards: Add a mixture of stable isotope-labeled internal standards (SIL-IS) at the beginning of extraction to correct for technical variability [1].
- LC-MS Analysis:
- Chromatography: Use UHPLC with a C18 column for small molecules or a reversed-phase column for peptides. A water-acetonitrile gradient (with 0.1% formic acid) is standard [2] [6].
- Mass Spectrometry: Operate a high-resolution mass spectrometer (e.g., Q-TOF, Orbitrap) in data-dependent acquisition (DDA) mode. Full MS1 scans are followed by fragmentation (MS2) of the most intense ions [65] [6].
- Data Processing:
- Use software (e.g., MS-DIAL, XCMS) for peak picking, alignment, and deconvolution.
- Perform statistical analysis (PCA, PLS-DA) to group samples and rank features by importance (VIP score) and significance (p-value) [6] [28].
- Annotate significant features using accurate mass, isotopic pattern, and MS/MS spectral matching to databases (e.g., HMDB, METLIN) [1].
Protocol 2: Targeted LC-MS/MS Validation of Candidate Biomarkers
- Objective: To precisely quantify a shortlist of candidate biomarkers in a large, independent cohort [65].
- Assay Development:
- Transition Selection: For each candidate, optimize precursor ion > product ion transitions using synthetic standards.
- Internal Standards: Procure or synthesize stable isotope-labeled analogs (e.g., ^13^C-, ^15^N-labeled) for each target as internal standards [63].
- LC-MS/MS Analysis:
- Chromatography: Optimize a fast, isocratic or shallow gradient for maximum separation and peak shape of the target panel.
- Mass Spectrometry: Use a triple quadrupole (QQQ) or Q-Orbitrap instrument in Multiple Reaction Monitoring (MRM) or Parallel Reaction Monitoring (PRM) mode. This provides superior sensitivity, specificity, and linear dynamic range for quantification [65] [3].
- Quantification & Validation:
- Generate a calibration curve using analyte standards spiked into a synthetic or pooled control matrix.
- Determine assay performance parameters: linearity, limit of detection (LOD), limit of quantification (LOQ), precision, and accuracy [65].
- Apply the validated assay to the sample cohort to confirm the diagnostic/prognostic power of the biomarker panel.
Visualizing the Complementary Multi-Platform Strategy
For comprehensive metabolome coverage, LC-MS and GC-MS are often used complementarily. This integrated approach is critical for natural product profiling and disease biomarker discovery [6] [28].
The Researcher's Toolkit: Essential Reagents & Materials
Successful LC-MS-based metabolomics requires carefully selected reagents and consumables to ensure reproducibility and accuracy [65] [1].
Table 4: Key Research Reagent Solutions for LC-MS Metabolomics
| Reagent/Material | Function & Specification | Critical Role in Workflow |
|---|---|---|
| Extraction Solvents | Methanol, Acetonitrile, Chloroform, Methyl tert-butyl ether (MTBE). LC-MS grade purity is essential to reduce background noise [1]. | Quenches metabolism and extracts metabolites from biological matrices. Choice of solvent system (e.g., biphasic methanol/chloroform/water) determines coverage of polar vs. non-polar metabolites [1]. |
| Stable Isotope-Labeled Internal Standards (SIL-IS) | ^13^C-, ^15^N-, or ^2^H-labeled versions of target analytes or class representatives (e.g., L-2-chlorophenylalanine) [28]. | Added at sample preparation start. Corrects for analyte loss during extraction, matrix effects during ionization, and instrument variability, enabling accurate quantification [63] [1]. |
| Chromatography Buffers & Additives | Formic Acid, Ammonium Acetate, Ammonium Hydroxide. LC-MS grade, typically at 0.1% concentration [6]. | Modifies mobile phase pH to improve ionization efficiency and chromatographic peak shape (e.g., acid for positive mode, base for negative mode ESI). |
| High-Abundance Protein Depletion Kits | Immunoaffinity columns for specific removal of albumin, IgG, etc., from serum/plasma [65]. | Reduces dynamic range and minimizes ion suppression, allowing detection of low-abundance protein biomarkers. |
| Quality Control (QC) Pool | A pooled sample created by combining equal aliquots of all study samples [1]. | Injected repeatedly throughout the analytical sequence to monitor instrument stability, assess reproducibility, and correct for signal drift during data processing. |
| UHPLC Columns | Reverse-phase (e.g., C18), hydrophilic interaction (HILIC), or specialized columns. Sub-2μm particle size for high resolution [2]. | Provides the critical separation of complex mixtures prior to MS detection, reducing ion suppression and increasing metabolite identifications. |
Overcoming Analytical Challenges in Natural Product Profiling
Managing Matrix Complexity and Ion Suppression in LC-MS
In the field of natural product metabolomics, the choice between Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS) is foundational, shaping the scope, reliability, and depth of research outcomes. LC-MS has become the predominant platform for its ability to analyze a vast range of non-volatile, polar, and thermally labile compounds—a category that encompasses the majority of natural products, from flavonoids and alkaloids to lipids and peptides [14] [15]. Its superior sensitivity, often cited at a level of 10⁻¹⁵ mol, allows for the detection of trace-level metabolites in complex biological matrices [14]. However, this very strength is coupled with a significant and persistent challenge: matrix effects and the resultant ion suppression.
Ion suppression is a phenomenon where co-eluting compounds from a sample matrix interfere with the ionization efficiency of target analytes in the mass spectrometer's ion source, leading to reduced, variable, or even completely obscured signals [66] [67]. This effect is particularly pronounced in electrospray ionization (ESI), the soft ionization technique most common in LC-MS, where competition for charge on droplet surfaces can drastically alter quantification accuracy [66] [68]. In contrast, GC-MS, which typically employs electron ionization (EI) in a high-vacuum environment, is far less susceptible to such matrix-related ionization interferences, offering more robust and reproducible quantitation for volatile compounds [68] [15]. The critical thesis for modern metabolomics research, therefore, is not a question of which technique is universally superior, but how to strategically leverage their complementary strengths while developing robust strategies to mitigate the inherent weaknesses of LC-MS, specifically its vulnerability to matrix complexity.
This comparison guide objectively evaluates the performance of LC-MS against GC-MS within this context. It provides experimental data and protocols focused on diagnosing, understanding, and correcting for matrix effects, framing them as an essential component of method development to ensure the generation of reliable, high-fidelity metabolomic data for natural product discovery and analysis.
Foundational Comparison: LC-MS vs. GC-MS for Metabolite Analysis
The operational principles of LC-MS and GC-MS dictate their respective applications, advantages, and limitations. A clear understanding of these fundamentals is necessary to contextualize the specific issue of matrix effects.
LC-MS separates compounds in a liquid phase using columns packed with a stationary phase. Analytes are separated based on their polarity and interaction with this phase and the liquid mobile phase [14]. The eluted compounds are then ionized at atmospheric pressure, most commonly via ESI or Atmospheric Pressure Chemical Ionization (APCI), before being introduced into the mass spectrometer [14] [68]. This process allows for the analysis of a broad spectrum of molecules without the need for volatilization, making it ideal for thermally unstable natural products.
GC-MS first vaporizes samples in a heated inlet. The gaseous analytes are carried by an inert gas through a long capillary column, where they are separated based on their boiling points and interactions with the column's stationary phase [14] [15]. Separation is followed by ionization via EI, which bombards molecules with high-energy electrons, typically causing extensive and reproducible fragmentation [14] [15].
The table below summarizes the core differences that guide platform selection.
Table 1: Foundational Comparison of LC-MS and GC-MS for Metabolomics
| Aspect | GC-MS | LC-MS (ESI) | Performance Implication for Natural Products |
|---|---|---|---|
| Ionization Method | Electron Impact (EI) - Hard ionization [14] [15]. | Electrospray (ESI) / APCI - Soft ionization [14] [68]. | EI provides rich, reproducible fragment spectra for library matching; ESI often yields intact molecular ions (e.g., [M+H]+) but is prone to suppression. |
| Analyte Suitability | Volatile, thermally stable compounds (typically <500 Da) [15]. Often requires derivatization for polar metabolites. | Broad range: non-volatile, polar, ionic, and thermally labile compounds [14] [15]. No derivatization needed for most. | LC-MS covers a much wider chemical space of natural products. GC-MS is limited to volatile fractions (e.g., essential oils, some fatty acids). |
| Sample Preparation | Often complex, involving derivatization (e.g., silylation, methoximation) to increase volatility [28] [15]. | Can be simpler (protein precipitation, SPE), but requires careful optimization to minimize matrix [28]. | Derivatization for GC-MS adds time, cost, and potential for error. LC-MS prep must be designed to reduce ion suppression agents. |
| Matrix Effects & Ion Suppression | Generally low. EI occurs in high vacuum after chromatographic separation [68]. | High prevalence. Occurs in the ion source due to competition from co-eluting matrix [66] [67]. | A major differentiator. GC-MS offers more reliable quantification; LC-MS requires active mitigation strategies for accurate bioanalysis. |
| Identification Power | Excellent. Large, standardized EI spectral libraries (e.g., NIST) enable high-confidence identification [15]. | Growing but less universal. Relies on accurate mass, MS/MS fragmentation, and retention time [15]. | GC-MS is superior for identifying known compounds in its domain. LC-MS is powerful for novel compound discovery and profiling. |
| Typical Sensitivity | High (~10⁻¹² mol) [14]. | Very High (~10⁻¹⁵ mol) [14]. | LC-MS can detect lower-abundance metabolites, but this advantage can be nullified by severe ion suppression. |
A direct illustration of their complementary nature comes from a 2024 study on lupus nephritis biomarkers, which utilized both platforms for serum metabolomics. The research identified 41 potential biomarkers, with each platform detecting distinct but overlapping sets of metabolites associated with different pathways, underscoring the power of an integrated approach [28].
Experimental Focus: Protocols for Diagnosing and Managing Matrix Effects
Given that ion suppression is a critical vulnerability of LC-MS, its systematic evaluation and management are non-negotiable for rigorous method development. The following experimental protocols, drawn from current research, provide a roadmap.
Protocol 1: The Post-Column Infusion Experiment (The Qualitative Diagnostic)
This classic experiment visually maps the regions of ion suppression or enhancement across the chromatographic run time [66].
Objective: To identify the retention time windows where matrix components affect ionization. Procedure:
- A solution containing the analyte(s) of interest is continuously infused post-column into the LC effluent at a constant rate using a syringe pump.
- A blank matrix extract (e.g., processed plasma, plant extract) is injected onto the LC column.
- The MS records the signal of the infused analyte in selected reaction monitoring (SRM) or single ion monitoring (SIM) mode over the entire chromatographic run.
Data Interpretation: A stable signal indicates no matrix effect. A dip in the signal indicates ion suppression, while a peak indicates ion enhancement, both occurring at the retention time where interfering matrix components elute [66]. This method is invaluable for troubleshooting and for adjusting chromatographic conditions to shift the analyte's retention time away from suppression zones.
Protocol 2: The Post-Extraction Spiking Method (The Quantitative Assessment)
This quantitative method is recommended by regulatory guidelines (e.g., EMA, ICH M10) to calculate the Matrix Factor (MF) and is integral to bioanalytical method validation [67].
Objective: To quantify the absolute and relative matrix effect on precision and accuracy. Procedure (based on Matuszewski's approach and recent implementations) [67]:
- Prepare three sets of samples for each analyte at low and high concentrations:
- Set A (Neat Solution): Analyte spiked into pure mobile phase or solvent.
- Set B (Post-extraction Spike): Blank matrix is carried through the entire sample preparation process. After extraction and reconstruction, the analyte is spiked into the cleaned matrix extract.
- Set C (Pre-extraction Spike): Analyte is spiked into the blank matrix before the sample preparation process and carried through all steps.
- Analyze all sets by LC-MS/MS. The peak area responses are used for calculation.
- Absolute Matrix Factor (MF) = Peak Area (Set B) / Peak Area (Set A). An MF of 1 indicates no effect; <1 indicates suppression; >1 indicates enhancement.
- Process Efficiency (PE) = Peak Area (Set C) / Peak Area (Set A). This reflects the combined loss from both matrix effect and recovery during sample prep.
- Recovery (RE) = Peak Area (Set C) / Peak Area (Set B). Isolates the efficiency of the extraction process itself.
Data Interpretation: The precision (CV%) of the MF across different lots of matrix (e.g., 6 individual plasma lots) is critical. A high CV indicates a variable "relative matrix effect" that cannot be compensated for by a stable isotope-labeled internal standard (SIL-IS), posing a major risk to assay reliability [67]. Acceptance criteria often require a CV < 15% for the IS-normalized MF.
Protocol 3: A Novel Compensation Strategy - Post-Column Infusion of Standards (PCIS)
Recent research has developed PCIS from a diagnostic tool into a correction strategy for untargeted metabolomics [69].
Objective: To select an optimal correction standard for each detected feature to compensate for matrix effects. Procedure [69]:
- Create an Artificial Matrix Effect (MEart): A mixture of compounds known to disrupt the ESI process (e.g., salts, ion-pairing agents) is post-column infused while analyzing standards.
- Test Candidate PCISs: Multiple stable-isotope labeled (SIL) standards are evaluated as potential PCISs. The PCIS whose infusion best stabilizes the signal of a given analyte during the MEart experiment is selected.
- Validate with Biological Matrix (MEbio): The selected PCIS is then used in experiments with actual biological samples. The study showed 89% agreement in PCIS selection between the artificial and biological matrix effects, validating the approach [69].
Data Interpretation: This strategy allows for feature-specific matrix effect correction in complex, untargeted analyses where isotopically labeled internal standards are not available for every metabolite, significantly improving data accuracy [69].
Diagram 1: Mechanism and Impact of Ion Suppression in LC-ESI-MS.
Diagram 2: Experimental Workflow for Quantifying Matrix Effects and Recovery.
The Scientist's Toolkit: Essential Reagents and Materials
Successful management of matrix complexity requires careful selection of reagents and materials at every step.
Table 2: Key Research Reagent Solutions for Managing Matrix Effects in LC-MS Metabolomics
| Item | Function & Rationale | Example from Protocols |
|---|---|---|
| Stable Isotope-Labeled Internal Standards (SIL-IS) | The gold standard for compensation. Co-elutes with the native analyte, experiences identical matrix effects and recovery losses, allowing for accurate ratio-based quantification [67]. | Used in the post-extraction spiking method to calculate IS-normalized Matrix Factor [67]. |
| Post-Column Infusion Standard Mix | A solution of compounds infused post-column to diagnose (PCIS experiment) or correct for (novel PCIS strategy) matrix effects in real-time [69] [66]. | A mix of SIL standards used to select the optimal compensator for each feature based on artificial matrix effect [69]. |
| Protein Precipitants (e.g., Methanol, Acetonitrile) | First-line cleanup to remove proteins, a major source of matrix interference. Cold organic solvent mixtures are commonly used [28]. | Methanol-acetonitrile (2:1 v/v) used for serum metabolomics to precipitate proteins before LC-MS analysis [28]. |
| Solid-Phase Extraction (SPE) Cartridges | Selective cleanup to remove specific classes of interferents (e.g., lipids, salts, pigments) and concentrate analytes, reducing overall matrix load. | Commonly used for lipidomics or targeted metabolite classes from plant or plasma extracts. |
| Volatile LC-MS Buffers (Ammonium Formate/Acetate) | Required for ESI compatibility. Non-volatile salts (e.g., phosphate) cause severe ion suppression and instrument contamination [68]. | Used in mobile phases for hydrophilic interaction liquid chromatography (HILIC) or reverse-phase chromatography of polar metabolites. |
| High-Purity Solvents (LC-MS Grade) | Minimizes chemical noise and background ions that can contribute to baseline suppression and interfere with low-abundance metabolite detection. | LC-MS grade methanol, acetonitrile, and water are essential for mobile phase and sample preparation [67]. |
Comparative Performance Analysis and Future Trajectory
The evolution of both LC-MS and GC-MS technologies continues to reshape their application landscapes. For LC-MS, market and innovation trends are directly focused on overcoming its traditional weaknesses.
Table 3: Performance Outlook and Technological Trends
| Dimension | GC-MS Trajectory | LC-MS Trajectory (Addressing Matrix Challenges) |
|---|---|---|
| Market & Adoption | Mature, stable market. Lower operational cost (gas vs. solvents) [15]. | Rapid growth (CAGR ~6.5-7.2%), projected to reach ~$4.5B by 2032 [70] [71]. Driven by pharmaceutical, biotech, and clinical diagnostics demand. |
| Instrument Innovation | Incremental improvements in sensitivity and speed. Growth of GCxGC for complex samples [14]. | Focus on Robustness and Throughput: New ion sources (e.g., "StayClean"), automated maintenance, and drier vacuum pumps reduce downtime [72]. Higher Resolution: Proliferation of Q-TOF and Orbitrap systems for better selectivity in complex matrices [70] [72]. |
| Workflow Integration | Stand-alone analysis. | Trend towards Automation & Integration: Systems like the Vanquish Neo with tandem direct injection workflows perform column equilibration offline to maximize throughput [72]. Automated sample prep integration is key. |
| Data Analysis | Reliant on established libraries. | Advanced Software for Mitigation: New CDS and data analysis tools incorporate features for monitoring system performance and detecting matrix-related anomalies [72]. AI/ML for automated feature detection and suppression flagging is emerging. |
| Strategic Role in Metabolomics | Remains the gold standard for volatile metabolome quantification due to robustness and reproducibility. | Becoming the central, high-sensitivity platform for broad discovery, with a strong emphasis on developing standardized mitigation protocols (like PCIS [69]) and integrated cleanup workflows to ensure data quality. |
The comparison between LC-MS and GC-MS for natural product metabolomics reveals a landscape defined by complementary strengths. GC-MS provides robust, reproducible quantitative data for a specific chemical domain with minimal concern for ion suppression. LC-MS, while vulnerable to matrix effects, offers unrivalled breadth and sensitivity for exploring the vast chemistry of natural products.
Therefore, the strategic approach for researchers must be context-aware and problem-focused:
- For Targeted Analysis of Known Volatiles: GC-MS is often the more reliable and cost-effective choice.
- For Untargeted Discovery or Analysis of Non-Volatiles: LC-MS is indispensable. However, its adoption must be coupled with a mandatory method validation step that includes quantitative assessment of matrix effects and process efficiency using protocols like the post-extraction spiking method.
- For Comprehensive Studies: The combined use of both platforms, as demonstrated in clinical biomarker discovery [28], provides the most complete metabolic picture.
- Invest in Mitigation: Allocate resources not just to the LC-MS instrument, but to the necessary tools: SIL-IS where possible, optimized sample preparation workflows, and chromatographic methods designed to separate analytes from matrix interferents.
The future of reliable natural product metabolomics lies not in avoiding the complexity of LC-MS, but in systematically managing it through rigorous experimental design, validation, and the adoption of innovative compensation strategies, ensuring that its profound sensitivity translates into accurate biological insight.
Diagram 3: Decision Workflow for Selecting GC-MS vs. LC-MS in Metabolomics.
Optimizing Derivatization Efficiency and Avoiding Artifacts in GC-MS
Within the broader methodological debate in natural product metabolomics—choosing between liquid chromatography-mass spectrometry (LC-MS) and gas chromatography-mass spectrometry (GC-MS)—the requirement for chemical derivatization stands as the most defining characteristic of the GC-MS workflow. LC-MS excels in analyzing non-volatile and thermally labile compounds with minimal sample preparation, often requiring only dilution and filtration [3]. In contrast, GC-MS offers superior chromatographic resolution, robust spectral libraries, and highly reproducible quantitative analysis, but its application to the polar, non-volatile metabolites typical of natural products necessitates a chemical transformation step [20] [73]. This derivatization, while powerful, introduces a critical bottleneck: it can be time-consuming, prone to introducing artifacts, and a source of quantitative error [74] [75]. Therefore, for researchers committed to leveraging the strengths of GC-MS, the optimization of derivatization protocols and a rigorous understanding of artifact formation are not mere technical details but fundamental prerequisites for generating reliable, high-quality metabolomic data. This guide objectively compares modern strategies to maximize derivatization efficiency and ensure analytical fidelity against the backdrop of LC-MS alternatives.
Optimizing Derivatization for Enhanced Throughput and Coverage
The traditional derivatization process for GC-MS is a multi-step, off-line procedure that often involves drying samples, adding reagents, heating, and subsequent injection, which can take over an hour and exposes analytes to potential degradation [75]. Recent methodological advances focus on streamlining this process to improve throughput and reproducibility.
Injection-Port Derivatization (IPD)
A significant innovation is Injection-Port Derivatization (IPD), which integrates the derivatization reaction into the GC injection sequence. In this approach, the underivatized sample extract and the derivatization reagent are co-injected into the hot GC inlet, where instantaneous reaction occurs just prior to chromatographic separation [75].
- Protocol Summary: An optimized IPD protocol for plasma metabolomics involves mixing the dried sample extract with a silylation reagent like N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA). This mixture is then injected into a GC injection port heated to 270–300°C. The residence time in the hot inlet is brief (seconds), but sufficient for complete reaction, immediately volatilizing the derivatives for transfer to the column [75].
- Performance Gains: Compared to conventional off-line methods, IPD drastically reduces sample preparation time from hours to minutes, minimizes manual handling, and reduces the risk of degradation of unstable derivatives. It has been validated for diverse metabolite classes, including organic acids, amino acids, and sugars [75].
Systematic Optimization of Conventional Derivatization
For applications where IPD is not suitable, systematic optimization of conventional derivatization remains essential. A recent study on soy sauce koji provides a model framework [73].
- Protocol Summary: The optimized method for non-volatile metabolites involved:
- Extraction: 80% methanol, ultrasonic treatment for 10 min, followed by shaking for 20 hours.
- Derivatization: 100 µL of dried extract was reacted with 120 µL of MSTFA at an elevated temperature.
- Key Parameters: The study meticulously optimized the sample-to-reagent ratio, reaction temperature, and reaction time to maximize the yield and stability of trimethylsilyl (TMS) derivatives for over 60 compounds [73].
- Performance Outcome: This optimized Der-GC/MS method demonstrated markedly superior coverage compared to nuclear magnetic resonance (NMR) analysis of the same samples, identifying 63 compounds versus 24 identified by NMR [73]. The table below summarizes the comparative metabolite coverage.
Table 1: Metabolite Coverage: Optimized Der-GC/MS vs. NMR [73]
| Analytical Technique | Total Compounds Identified | Amino Acids | Organic Acids | Sugars/Sugar Alcohols | Fatty Acids |
|---|---|---|---|---|---|
| Der-GC/MS | 63 | 17 | 12 | 18 | 4 |
| NMR | 24 | 13 | 4 | 4 | 0 |
Identifying and Preventing Analytical Artifacts
Artifacts—compounds generated during sample preparation and analysis rather than present in the original biological sample—pose a significant threat to data integrity in both GC-MS and LC-MS. The sources differ between the two platforms.
Artifacts in GC-MS: Derivatization and Solvent Reactions
In GC-MS, artifacts predominantly arise from the derivatization chemistry itself and from unwanted reactions between analytes and extraction solvents [76].
- Common Artifact Pathways:
- Transesterification: In extracts containing esters (e.g., glycerolipids), alcohols like methanol or ethanol can catalyze the exchange of alkyl groups, creating new methyl or ethyl esters that misrepresent the native lipid profile [76].
- Esterification: Free carboxylic acids (e.g., fatty acids, organic acids) can react with alcohol solvents to form alkyl esters. For instance, using methanol can lead to the formation of methyl ester artifacts [76].
- Degradation under Heat: Thermally labile compounds can decompose in the hot GC inlet or column, producing breakdown products mistaken for genuine metabolites. Incomplete derivatization can also yield multiple derivatives from a single analyte, complicating quantification [75].
- Prevention Strategies:
- Control Reaction Conditions: Precisely control derivatization time and temperature to minimize side reactions and ensure completeness.
- Solvent Selection: Be aware of solvent-analyte interactions. Avoid storing samples in reactive solvents like chloroform, which can promote oxidations [76]. Use high-purity solvents to minimize contaminant-driven reactions.
- Use Deuterated Reagents: Employing deuterated derivatization reagents (e.g., d₉-MSTFA) or extraction solvents can help identify artifact peaks in mass spectra, as they introduce a predictable mass shift.
- Analyze Underivatized Extracts: Where possible, analyze a portion of the sample via LC-MS to distinguish native compounds from GC-specific derivatization artifacts.
The following diagram illustrates the primary pathways for artifact formation in GC-MS sample preparation.
Artifacts in LC-MS: Adduct and Cluster Formation
LC-MS artifacts are primarily formed during the ionization process, especially in electrospray ionization (ESI) [77].
- Common Artifact Pathways:
- Adduct Formation: Analyte molecules ([M]) can combine with ions ubiquitous in solvents and samples (Na⁺, K⁺, NH₄⁺, formate) to form signals for [M+Na]⁺, [M+K]⁺, etc. These adducts split the signal of a single compound across multiple m/z values, reducing sensitivity and complicating spectra [77].
- Cluster/Dimer Formation: Molecules can form multimers (e.g., [2M+H]⁺) in the electrospray droplets, which are detected as higher m/z ions [77].
- In-Source Fragmentation: Some labile compounds may fragment before the first mass analyzer, producing a signal for the fragment that cannot be distinguished from a genuine low-mass metabolite.
- Prevention and Management:
- Mobile Phase Additives: Using consistent, high-purity buffers and additives can help control and standardize adduct formation.
- Clean Sample Preparation: SPE cleanup or protein precipitation reduces matrix ions that promote adduct formation [78].
- Data Analysis Tools: Software can recognize and account for common adducts and dimers during peak alignment and annotation, collapsing related signals into a single feature.
Performance Comparison: GC-MS vs. LC-MS in Metabolomics
The choice between GC-MS and LC-MS involves trade-offs across several performance metrics, heavily influenced by the derivatization requirement of GC-MS.
Sensitivity and Limit of Detection (LOD)
Derivatization in GC-MS generally enhances volatility and can improve ionization efficiency in the electron impact (EI) source, often leading to excellent sensitivity. For example, in the analysis of resin acids, GC-MS achieved limits of detection below 0.2 µg/L, outperforming LC-APCI-MS (<3 µg/L) [74]. However, for compounds that ionize exceptionally well under ESI (e.g., pre-charged molecules), LC-MS may demonstrate superior sensitivity without the need for derivatization.
Metabolite Coverage and Complementarity
The techniques are highly complementary. A direct comparison in a clinical metabolomics study on lupus nephritis serum samples revealed non-overlapping coverage [28].
Table 2: Complementary Metabolite Coverage in Serum Metabolomics [28]
| Analytical Platform | Total Metabolites Detected | Key Classes of Unique Metabolites | Primary Reason for Coverage |
|---|---|---|---|
| GC-MS | 28 | Organic acids, sugars, amino acids, fatty acids | Derivatization makes polar, non-volatile metabolites amenable to GC analysis. |
| LC-MS (RP) | 13 | Phospholipids, bile acids, complex lipids | Native analysis of medium-to-low polarity molecules. |
This complementary nature means that a multi-platform approach (GC-MS + LC-MS) provides the most comprehensive snapshot of the metabolome [20] [28].
Reproducibility, Throughput, and Ease of Use
- Reproducibility: GC-MS with EI ionization benefits from highly reproducible, instrument-independent mass spectra, facilitating library matching. Derivatization is a major source of variability in GC-MS that must be tightly controlled. LC-MS reproducibility can be affected by matrix effects and fluctuating ionization efficiency, often necessitating isotope-labeled internal standards for quantification [78].
- Throughput: LC-MS typically has a throughput advantage for broad screens as it usually requires less extensive sample preparation—often just protein precipitation or dilution [78] [3]. The derivatization step in GC-MS adds significant time, though methods like IPD are closing this gap [75].
- Structural Information: GC-EI-MS provides rich, fragment-rich spectra ideal for identifying unknown compounds via library search. LC-MS/MS (tandem MS) provides precursor-product ion relationships that are very useful for structural elucidation and targeted quantification.
The workflow diagram below summarizes the key decision points and steps when choosing and applying GC-MS with derivatization.
The Scientist's Toolkit: Essential Reagents and Materials
Successful and artifact-minimized GC-MS metabolomics relies on specific, high-purity reagents.
Table 3: Key Reagent Solutions for Derivatization GC-MS Metabolomics
| Reagent/Material | Function | Key Consideration for Avoiding Artifacts |
|---|---|---|
| MSTFA (N-Methyl-N-(trimethylsilyl)trifluoroacetamide) | Primary silylation reagent for -OH, -COOH, -NH groups. | Use fresh, anhydrous reagent; contains own catalyst; preferred for IPD [75] [73]. |
| BSTFA (BSTFA with 1% TMCS) | Silylation reagent. TMCS acts as a catalyst. | Ensure dryness of sample; TMCS can be too reactive for some compounds [74] [28]. |
| Methoxyamine Hydrochloride (in Pyridine) | Performs oximation of carbonyl groups (ketones, aldehydes) before silylation. | Prevents multiple derivative peaks from reducing sugars; pyridine must be anhydrous [28]. |
| Anhydrous Pyridine | Common solvent for derivatization reactions. Serves as a base and moisture scavenger. | Critical: Must be kept anhydrous. Any water leads to incomplete derivatization and reagent hydrolysis [76]. |
| High-Purity, Anhydrous Methanol & Acetonitrile | Extraction and reconstitution solvents. | Use LC-MS grade. Trace water or acids in methanol can cause transesterification or esterification artifacts [76]. |
| Deuterated Derivatization Standards (e.g., d₉-MSTFA) | Internal standards for derivatization control or artifact identification. | Adds a predictable 9 Da mass shift per TMS group, helping to distinguish artifact peaks in complex spectra. |
| Inert GC Inlet Liners (e.g., deactivated glass wool) | For injection-port derivatization (IPD) or high-risk samples. | Minimizes catalytic degradation of sensitive analytes in the hot injection port. |
In the context of natural product metabolomics, GC-MS remains a powerhouse for the quantitative and reproducible analysis of primary metabolites, albeit with a mandatory derivatization step. The evolution of strategies like Injection-Port Derivatization addresses historical throughput limitations [75], while a rigorous, chemistry-driven understanding of artifact formation pathways—from solvent interactions to thermal degradation—is essential for data fidelity [76]. When compared directly, GC-MS and LC-MS show complementary strengths in coverage and sensitivity [74] [28]. The optimal choice is not always singular; a well-designed, multi-platform approach often provides the most comprehensive and reliable metabolomic profile. For researchers selecting GC-MS, investing time in optimizing and controlling the derivatization process is the definitive factor in unlocking the technique's full potential while ensuring the biological validity of the results.
The field of natural product metabolomics and drug discovery is fundamentally constrained by the speed and reliability of sample preparation and analysis. The shift from traditional one-variable-at-a-time experimentation to high-throughput experimentation (HTE) enables the parallel processing of dozens to hundreds of samples, dramatically accelerating data generation for applications ranging from therapeutic drug monitoring to biomarker discovery [79]. Within this paradigm, method automation and the standardized 96-well plate format have emerged as critical pillars for achieving scalability, reproducibility, and efficiency.
This guide is framed within a broader thesis investigating LC-MS versus GC-MS for natural product research. While both techniques are powerful, their integration with automated, high-throughput workflows dictates their applicability in modern laboratories. LC-MS is generally favored for its broad compatibility with non-volatile and thermally labile metabolites prevalent in natural products, whereas GC-MS offers superior resolution for volatile compounds but often requires extensive derivatization [80] [81]. The convergence of automated liquid handling, parallel extraction technologies, and 96-well formatted mass spectrometry is dissolving previous bottlenecks, enabling robust comparative metabolomics at a scale previously unattainable. This guide objectively compares the performance of automated, plate-based methods against traditional or manual approaches, supported by recent experimental data.
Quantitative Comparison of Automated High-Throughput Platforms
The following tables summarize the performance characteristics of recent automated platforms, highlighting gains in throughput, precision, and cost-effectiveness.
Table 1: Performance Comparison of Automated High-Throughput Analytical Workflows
| Application & Platform | Throughput (Samples/Day) | Key Performance Metrics | Comparative Advantage | Primary Reference |
|---|---|---|---|---|
| LC-MS/MS for CBD TDM (Automated PP) | ~96-192 (est. based on plate format) | Intraday precision: 1.7-4.5%; Accuracy: 95.6-104.4%; Strong correlation with manual method (Passing-Bablok regression) [82] [83]. | Full automation eliminates manual steps (solvent add, mix, centrifuge, transfer); superior reproducibility for clinical TDM [83]. | Palmisani et al. (2025) [82] [83] |
| Electro-Extraction (EE) for Acylcarnitines (Fully Automated EE-LC-MS) | 120 | Extraction recovery: Up to 99%; Enrichment factor: Up to 400; Cost: <€0.10 per sample [84]. | Exceptional recovery and enrichment for low-abundance analytes; highly cost-effective for large-scale studies [84]. | Sciencedirect (2025) [84] |
| Dual LC-MS/MS & GC-MS/MS for Smoke Biomarkers (96-well SPE) | High-throughput (exact number not specified) | Comprehensive profiling of NNAL metabolites and PheT; enabled large-scale epidemiologic study (N > 2600) [80]. | Single 96-well platform for both LC (polar metabolites) and GC (silylated metabolites) workflows maximizes laboratory efficiency [80]. | Hecht et al. (2013) [80] |
| Protein Precipitation Filtration vs. Pellet Method (96-well Filter Plate) | ~2-4x faster than pelleting | Overall %RSD: 5.7% (Filter) vs. 7.5% (Pellet); Reduced variability at low conc.; eliminated supernatant pipetting [85]. | Simplified workflow reduces manual error and improves precision, especially in drug discovery screening [85]. | Chromatography Online (2006) [85] |
Table 2: Suitability of High-Throughput Methods for Natural Product Metabolomics
| Method/Technique | Best Suited For | Compatibility with LC-MS | Compatibility with GC-MS | Key Considerations for Natural Products |
|---|---|---|---|---|
| Automated Protein Precipitation (PP) | Primary metabolite screening, therapeutic drug monitoring [83]. | Excellent; direct analysis of supernatant [85] [83]. | Poor; requires extract drying/derivatization. | Fast and simple for crude extracts; may leave complex matrix for LC-MS to resolve [81]. |
| Microextraction in 96-well Plates (SPME/LPME) | Green chemistry; targeted analysis of specific analyte classes [86]. | Good for LC-MS after analyte desorption. | Excellent for GC-MS; direct thermal desorption possible. | Minimizes solvent use; high enrichment factors ideal for low-concentration biomarkers [86] [84]. |
| Automated Solid-Phase Extraction (SPE) | Purification and concentration of complex mixtures [80]. | Excellent; various stationary phases available. | Good after eluent evaporation and derivatization. | Essential for removing interfering compounds in plant extracts; can be automated in 96-well format [80]. |
| Supported Liquid Extraction (SLE) | Efficient recovery of analytes from biological fluids [80]. | Excellent. | Good after derivatization. | Often used in biomarker studies; provides clean extracts for sensitive analysis [80]. |
Detailed Experimental Protocols from Featured Studies
This protocol describes the automated sample preparation for Cannabidiol (CBD) and its metabolite 7-hydroxy-CBD in human serum.
- Sample Preparation: Implemented on an integrated robotic platform. Briefly, 50 µL of serum was aliquoted into a 96-well protein precipitation plate. The platform automatically added 150 µL of acetonitrile containing the internal standard (CBD-d3), mixed, centrifuged, and filtered the supernatant through a PTFE membrane into a fresh 96-well collection plate.
- LC-MS/MS Analysis: Chromatography used a C18 column with a gradient of water and acetonitrile (both with 0.1% formic acid). Detection was via tandem mass spectrometry in positive electrospray ionization (ESI+) mode using multiple reaction monitoring (MRM).
- Validation: The method was validated per EMA guidelines. Calibration curves were linear (R² > 0.99). Precision (CV) was <15% at the LLOQ and <10% at other levels, with accuracy between 85-115% [83].
This protocol outlines a novel, fully automated electro-extraction for polar metabolites from plasma and tissue.
- Electro-Extraction: A CTC PAL3 autosampler was integrated with a custom EE device. For plasma, 20 µL of sample was loaded. An electric field was applied, migrating charged acylcarnitines from the sample through a supported liquid membrane (SLM) and into an acceptor solution.
- Optimization: Parameters (voltage, pH, time) were optimized via Design of Experiment (DoE), achieving near-complete (>95%) recovery of model acylcarnitines.
- Downstream Analysis: The acceptor solution was directly injected into an LC-MS/MS system for quantification. The end-to-end workflow processed 120 samples per day at a consumables cost below €0.10 per sample [84].
This protocol details a high-throughput 96-well method for two distinct classes of biomarkers.
- For NNAL (LC-MS/MS Analysis): Urine aliquots were processed in three parallel 96-well plates for "free," "free + N-glucuronide," and "total" NNAL. Samples underwent supported liquid extraction (SLE) followed by mixed-mode cation-exchange SPE. Extracts were analyzed by LC-MS/MS.
- For PheT (GC-MS/MS Analysis): Separate urine aliquots were cleaned up via styrene-divinylbenzene SPE in a 96-well plate. The eluent was derivatized to form trimethylsilyl (TMS) ethers and analyzed by GC-MS/MS.
- Throughput: This parallel, plate-based approach enabled the processing and analysis of thousands of samples for a large molecular epidemiology study [80].
Visualizing High-Throughput Workflows
Diagram 1: Automated vs. Manual Workflow for TDM
Diagram 2: Parallel LC-MS & GC-MS Workflow for Biomarkers
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful implementation of high-throughput, automated metabolomics relies on specialized materials and reagents. The following toolkit details essential components derived from the featured studies.
Table 3: Essential Toolkit for Automated High-Throughput Metabolomics
| Category | Specific Item/Example | Function in Workflow | Key Benefit/Consideration |
|---|---|---|---|
| Sample Preparation & Plate Hardware | 96-well Protein Precipitation Filtration Plates (e.g., PTFE membrane) [85] | Combines protein precipitation and filtration in one step; eliminates manual supernatant transfer. | Critical for automation: Enables centrifugal-driven filtration directly into an analysis plate, drastically improving reproducibility and speed [85]. |
| 96-well Solid Phase Extraction (SPE) Plates (e.g., SLE+, Mixed-Mode Cation Exchange) [80] | Provides selective cleanup and concentration of analytes from complex matrices like urine or plant extracts. | Allows parallel processing of 96 samples; choice of sorbent (C18, MCX, polymer) is analyte-dependent [86] [80]. | |
| Deep-well (1-2 mL) 96-well Plates | Used for sample incubation, extraction, and storage in workflows like microsomal stability assays [87]. | Provides sufficient volume for multi-step reactions and quenching, compatible with automated liquid handlers. | |
| Automation & Liquid Handling | Robotic Liquid Handler / Autosampler (e.g., CTC PAL3 series) [84] [87] | Performs precise, unattended liquid transfers, dilutions, and injections. Can integrate with other modules (centrifuge, incubator). | Core of automation: Enables walk-away sample prep and injection. The open-bed design offers flexibility for custom workflows [87]. |
| Automated Centrifuge with 96-well plate rotors | Provides consistent pelleting or filtration force for precipitation and SPE steps within an automated sequence. | Eliminates a major manual bottleneck and variability source in sample preparation [83]. | |
| Analytical Standards & Reagents | Stable Isotope-Labeled Internal Standards (e.g., CBD-d3, ¹³C₆-NNAL) [83] [80] | Added at the very beginning of sample prep to correct for losses during extraction and matrix effects during MS analysis. | Essential for quantitative accuracy, especially in complex biological matrices. Must be chemically identical to the analyte. |
| High-Purity, LC-MS Grade Solvents | Used for extraction, mobile phases, and system washing. | Minimizes background noise and ion suppression in mass spectrometry, ensuring sensitivity and robust performance. | |
| Chromatography | UPLC/HPLC Columns for Fast Separations (e.g., C18, T3) [87] | Provides high-resolution separation of metabolites prior to MS detection. Short, small-particle columns enable fast gradients. | Increases throughput: Run times of 2-5 minutes per sample are common in high-throughput ADME and TDM assays [87]. |
| Data Processing | Automated Data Processing Software (e.g., QuanOptimize) [87] | Automates method optimization, peak integration, calibration curve generation, and report creation. | Manages the high data volume from HTE, reduces manual review time, and minimizes human error in data handling [79] [87]. |
The integration of full automation, 96-well plate formats, and advanced mass spectrometry represents the current zenith of high-throughput capability for natural product metabolomics and bioanalysis. As evidenced by the comparative data, automated workflows consistently deliver superior precision, higher throughput, and better scalability than their manual counterparts, while simultaneously reducing operational costs [84] [83].
The choice between LC-MS and GC-MS within these automated paradigms remains context-dependent. LC-MS is the more versatile backbone for high-throughput workflows, seamlessly interfacing with automated liquid-based sample prep and excelling in the analysis of a broad spectrum of natural products [83] [87]. GC-MS remains indispensable for volatile compounds and requires its own optimized, automated derivatization and injection sequences, often running in parallel to LC-MS workflows as shown in biomarker studies [80] [81].
The future direction points toward even greater integration and intelligence. Advances include: 1) Ultra-HTE using 1536-well plates [79], 2) AI-driven workflow optimization and data analysis [79] [87], and 3) Multi-omics integration on single automated platforms. For researchers, the strategic investment in modular automation and standardized plate-based protocols is no longer a luxury but a necessity to maintain competitiveness, ensure data integrity, and unlock the full potential of metabolomics in drug development and natural product research.
Systematic Optimization Using Design of Experiment (DoE) Strategies
This comparison guide objectively evaluates the performance of Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS) within the context of natural product metabolomics research. Framed within a broader thesis on analytical platform selection, the guide employs principles of Design of Experiments (DoE) to systematically compare the techniques across critical parameters including metabolite coverage, sensitivity, and suitability for complex plant extracts. Supporting experimental data, derived from published metabolomics studies, are summarized in structured tables. Detailed DoE-optimized protocols and visual workflow diagrams are provided to equip researchers and drug development professionals with a rigorous framework for method selection and optimization in the discovery of bioactive natural compounds.
Natural products represent a vital source for drug discovery, but their complex chemical composition presents a significant analytical challenge [18]. Metabolomics, the comprehensive study of small molecules, has emerged as an indispensable tool for profiling the hundreds to thousands of metabolites in plant extracts, moving beyond classical bioassay-guided fractionation [18] [88]. The core analytical platforms in this field are LC-MS and GC-MS, each with distinct advantages and limitations. LC-MS is renowned for its high sensitivity and ability to directly analyze a broad spectrum of nonvolatile, polar, and thermally labile compounds without derivation [2]. Conversely, GC-MS, particularly when enhanced by two-dimensional techniques (GC×GC-MS), offers superior chromatographic resolution and robust, reproducible spectral libraries, but primarily for volatile or chemically derivatized metabolites [89]. The choice between these platforms profoundly influences the depth and reliability of metabolomic data. This guide utilizes a systematic DoE framework—a statistical approach for optimizing processes by simultaneously evaluating multiple factors—to compare their performance objectively. Applying DoE principles moves beyond a one-variable-at-a-time (OVAT) comparison, allowing for a more efficient and insightful analysis of how different technical factors interact to affect outcomes in natural product analysis [90] [91].
Foundational Concepts: DoE in Analytical Method Optimization
Design of Experiments is a systematic methodology used to plan, conduct, and analyze controlled tests to evaluate the factors that influence a process or response. In contrast to the traditional OVAT approach, which is inefficient and incapable of detecting interactions between factors, DoE allows for the simultaneous variation of multiple parameters (e.g., column temperature, gradient time, ionization voltage) to build a predictive model of the analytical system [90] [91].
- Key DoE Designs:
- Screening Designs (e.g., Fractional Factorial): Used to identify the most influential factors from a large set with a minimal number of experimental runs.
- Response Surface Methodology (RSM) Designs (e.g., Central Composite, Box-Behnken): Used to model curvature and find the optimal settings for critical factors after they have been identified. Central composite designs are noted for their strong performance in multi-objective optimization of complex systems [92].
- Application to Metabolomics: In metabolomics, DoE is crucial for optimizing complex, multi-step workflows including sample extraction, chromatography, and ionization to maximize metabolite coverage and signal response while ensuring robustness [93] [91]. Its implementation helps save time and resources while providing reliable, comprehensive results that precisely represent the experimental design space [91].
Platform Performance Comparison via DoE Framework
A DoE-style comparison evaluates LC-MS and GC-MS across several interconnected response variables. The following tables synthesize experimental data from metabolomics studies to compare their performance.
Table 1: Comparison of Fundamental Analytical Characteristics
| Characteristic | LC-MS (ESI/APCI) | GC-MS (EI after Derivatization) | Experimental Context & DoE Insight |
|---|---|---|---|
| Analyte Coverage | Broad: Non-volatile, polar, high molecular weight (e.g., flavonoids, saponins, peptides). | Narrower: Volatile, thermally stable, or derivatizable compounds (e.g., sugars, organic acids, fatty acids, essential oils). | LC-MS covers complementary chemical space to GC-MS. A DoE screening design on sample preparation can determine the optimal solvent system (e.g., MeOH/CHCl₃/H₂O ratios) for broad coverage [1]. |
| Sensitivity | High (fg-pg levels); excels for polar molecules in complex matrices. | Very High (fg levels); excellent for volatile/small molecules. | Sensitivity is factor-dependent. A DoE on ionization parameters (e.g., gas temp, flow) in LC-MS can optimize signal-to-noise for trace natural products [2]. |
| Chromatographic Resolution | Good to Very Good (with UHPLC). | Excellent (especially with GC×GC). | GC×GC provides superior peak capacity. A DoE on GC temperature programming and column selection is key for separating complex essential oil profiles [89]. |
| Structural Elucidation | Provides molecular ion & fragment patterns (MS/MS); library matching less universal. | Provides reproducible, library-searchable electron ionization (EI) fragmentation patterns. | GC-MS EI spectra are highly standardized. For LC-MS, a DoE on collision energy can optimize MS/MS spectra quality for novel compound annotation [2]. |
| Sample Throughput | High; minimal preparation for many compounds. | Lower; often requires lengthy derivatization (e.g., methoximation, silylation). | Derivatization is a critical sample prep factor. A DoE can optimize derivatization time and temperature to maximize response for key metabolite classes [91]. |
| Quantitation Robustness | Good; can be affected by matrix-induced ionization suppression. | Excellent; stable EI ionization is less matrix-sensitive. | LC-MS quantitation benefits from DoE-optimized use of internal standards (e.g., isotope-labeled) to correct for matrix effects [1]. |
Table 2: Performance in Natural Product Metabolomics Case Studies
| Study Focus | LC-MS Findings | GC-MS/GC×GC-MS Findings | DoE-Optimized Parameter |
|---|---|---|---|
| Tea (Camellia sinensis) Metabolome [89] | Ideal for profiling catechins, theaflavins, and other polar polyphenols without derivation. | GC×GC-MS identified ~200 metabolites, including volatiles; tracked seasonal changes in flavor compounds. Profiling required comprehensive sample preparation. | Sample Introduction: DoE for SPME or SBSE fiber selection, time, and temperature to capture representative volatile profiles. |
| Plant Extract Drug Discovery [18] [88] | Indispensable for untargeted profiling of crude extracts; directly detects most secondary metabolites. | Can miss >50% of compounds in a complex natural product sample if not volatile [89]. | Extraction Solvent: A multi-factor DoE (solvent type, ratio, time) is essential to maximize metabolite range for LC-MS analysis [18] [1]. |
| Metabolite Identification | Relies on accurate mass, isotope patterns, and MS/MS; enhanced by molecular networking tools (e.g., GNPS). | Powerful for matching against extensive, standardized EI mass spectral libraries. | Data Acquisition: A DoE for LC-MS/MS data-dependent acquisition (DDA) settings (isolation width, collision energy ramp) improves MS/MS coverage. |
Detailed DoE-Optimized Experimental Protocols
Protocol 1: DoE for Optimizing Biphasic Extraction of Plant Metabolites for LC-MS Analysis This protocol uses a Response Surface Methodology (Box-Behnken or Central Composite Design) to optimize the yield and diversity of metabolites extracted from plant tissue.
- Define Objective & Responses: Maximize total ion chromatogram (TIC) peak count and area for key metabolite classes (e.g., phenolics, alkaloids).
- Select Critical Factors and Ranges:
- Factor A: Methanol:Water ratio (e.g., 60:40 to 90:10 v/v).
- Factor B: Extraction time with sonication (e.g., 5 to 30 minutes).
- Factor C: Solvent-to-sample ratio (e.g., 10:1 to 50:1 v/w).
- Execute DoE Runs: Generate and randomize the experimental run order from the design matrix. For each run, homogenize frozen plant tissue (e.g., 50 mg) with the specified solvent mixture, sonicate, centrifuge, and collect supernatant.
- Analysis & Model Building: Analyze all extracts using a standardized LC-MS method. Integrate TIC and responses. Use statistical software to fit a quadratic model and generate 3D response surfaces.
- Validation: Predict the optimal factor settings from the model. Prepare and analyze triplicate validation extracts at these conditions. Confirm that the measured responses fall within the model's prediction intervals [18] [91] [1].
Protocol 2: DoE for Optimizing GC-MS Derivatization of Polar Metabolites from a Herbal Product This protocol employs a factorial design to optimize the two-step derivatization (methoximation followed by silylation) required for GC-MS analysis of sugars and organic acids.
- Define Objective & Responses: Maximize peak response and shape for a panel of target polar metabolites.
- Select Factors and Levels:
- Factor A: Oximation time (e.g., 60 vs. 90 minutes).
- Factor B: Silylation temperature (e.g., 70°C vs. 80°C).
- Factor C: Silylation time (e.g., 30 vs. 60 minutes).
- Execute DoE Runs: Following a full or fractional factorial design, derivatize aliquots of a standardized herbal extract. Use a robotic autosampler for precision.
- Analysis & Interpretation: Analyze all samples by GC-MS. Measure peak height/area for key metabolites. Perform Analysis of Variance (ANOVA) to identify significant main effects and interaction effects (e.g., does the optimal silylation time depend on temperature?).
- Optimum Selection: Identify the factor level combination that yields the highest overall response across the target metabolite panel [91].
Visualization of Workflows and Logical Relationships
Title: DoE-Driven Metabolomics Workflow for Platform Selection
Title: Metabolite Class Coverage of LC-MS vs. GC-MS
The Scientist's Toolkit: Essential Research Reagents & Materials
The following table details key consumables and reagents critical for conducting DoE-optimized metabolomics studies of natural products.
Table 3: Research Reagent Solutions for Natural Product Metabolomics
| Item/Category | Function in Workflow | DoE Optimization Context | Primary Platform |
|---|---|---|---|
| Methanol (MeOH), LC-MS Grade | Primary extraction solvent for polar metabolites; mobile phase component. | Factor in solvent system optimization (e.g., MeOH:H₂O or MeOH:CHCl₃ ratios) to maximize metabolite recovery [1]. | Primarily LC-MS |
| Chloroform (CHCl₃) or Methyl tert-Butyl Ether (MTBE) | Organic solvent for biphasic extraction; partitions non-polar lipids and metabolites. | Safer alternative to CHCl₃. Factor in optimizing biphasic separation for lipidomics or comprehensive metabolomics [18] [1]. | LC-MS (Lipidomics) |
| Derivatization Reagents:• MSTFA (N-Methyl-N-(trimethylsilyl)trifluoroacetamide)• Methoxyamine hydrochloride | Chemical modification of polar functional groups (-OH, -COOH, -NH₂) to make metabolites volatile and thermally stable for GC-MS. | Critical Factors: Derivatization time, temperature, and reagent volume are key variables in a GC-MS sample prep DoE [91]. | GC-MS |
| Stable Isotope-Labeled Internal Standards(e.g., ¹³C, ¹⁵N labeled amino acids, fatty acids) | Added at known concentration pre-extraction to correct for analyte loss, matrix effects, and instrument variability. | Essential for robust quantification. Selection of appropriate class-specific standards is a foundational QA step before DoE [1]. | LC-MS & GC-MS |
| Solid-Phase Microextraction (SPME) Fibers | Solventless extraction and preconcentration of volatile organic compounds (VOCs) from headspace. | Fiber coating type, extraction time, and temperature are factors in a DoE for profiling plant volatiles via GC-MS [89]. | GC-MS |
| Quality Control (QC) Pooled Sample | A representative pool of all study samples analyzed repeatedly throughout the batch to monitor instrument stability and data reproducibility. | Not a DoE factor, but a critical QA material. Its consistency is used to assess the performance of the DoE-optimized method over time [1]. | LC-MS & GC-MS |
The systematic comparison facilitated by a DoE framework reveals that LC-MS and GC-MS are complementary, not competing, platforms in natural product metabolomics. LC-MS is the primary workhorse for untargeted discovery and profiling of the majority of secondary metabolites due to its broad analyte coverage and minimal sample preparation requirements [2] [88]. GC-MS (especially GC×GC-MS) is the specialist tool of choice for volatile metabolomics, offering unrivaled resolution and identification confidence for essential oils and central metabolites after derivatization [89].
Strategic recommendations for researchers are:
- Lead with LC-MS: For initial, comprehensive fingerprinting of unknown plant extracts or bioactivity-guided studies, LC-MS should be the first-line platform.
- Employ GC-MS for Specific Targets: Incorporate GC-MS when the research question specifically involves volatiles, flavors, aromas, or core metabolic pathway intermediates (e.g., in plant physiology studies).
- Adopt a DoE Mindset: Systematically optimize critical steps—especially sample preparation—using screening and response surface designs. This is more efficient than OVAT and reveals critical factor interactions, leading to more robust and reproducible methods [93] [91].
- Consider Data Integration: For a complete systems biology understanding, integrate data from both platforms. Computational metabolomics and molecular networking tools are increasingly facilitating this synthesis, offering a more holistic view of the plant metabolome and its bioactivity [94].
The future of natural product metabolomics lies in the intelligent, question-driven combination of these platforms, underpinned by rigorous, DoE-informed methodology to ensure data quality and accelerate discovery.
Natural products (NPs) represent a vast and chemically diverse reservoir of bioactive compounds crucial for drug discovery and development. However, this complexity poses a significant analytical challenge for metabolomics, the comprehensive study of small-molecule metabolites [1]. Reproducible and accurate metabolite profiling is the cornerstone of valid biological interpretation, yet it is threatened by inherent variability in sample preparation, instrument performance, and data acquisition [1] [95].
Within this context, Gas Chromatography-Mass Spectrometry (GC-MS) and Liquid Chromatography-Mass Spectrometry (LC-MS) have emerged as the two dominant analytical platforms. Their orthogonal separation mechanisms make them differentially suited for specific chemical classes within NP extracts [15] [32]. Consequently, the choice between—or integration of—these techniques is a fundamental methodological decision that directly impacts data quality and reproducibility [32].
This guide provides a comparative framework for selecting and optimizing GC-MS and LC-MS workflows in NP metabolomics. It objectively evaluates their performance, underscores the non-negotiable role of internal standards (IS) and systematic quality control (QC), and provides actionable protocols to ensure data integrity from sample collection to statistical analysis.
Platform Comparison: GC-MS versus LC-MS for Natural Product Metabolomics
The core distinction between GC-MS and LC-MS lies in their separation principles, which dictate the types of metabolites they can optimally analyze. The following table summarizes their key characteristics and performance metrics, which are critical for platform selection [15] [32] [3].
Table 1: Comparative Performance of GC-MS and LC-MS for Natural Product Metabolomics
| Criterion | GC-MS | LC-MS |
|---|---|---|
| Core Principle | Separation by volatility and gas-phase interaction with a stationary phase. | Separation by polarity/solubility in liquid mobile phase interacting with a stationary phase [15]. |
| Ideal Analyte Profile | Volatile, semi-volatile, and thermally stable compounds (typically <500 Da). Examples: Essential oils, fatty acids, organic acids, sugars (after derivatization), certain alkaloids [15] [3]. | Non-volatile, thermally labile, polar, and high molecular weight compounds. Examples: Flavonoids, glycosides, peptides, polar lipids, most pharmaceuticals [15] [3]. |
| Typical Ionization | Electron Ionization (EI) - a "hard" method producing extensive, reproducible fragmentation [15]. | Electrospray Ionization (ESI) or Atmospheric Pressure Chemical Ionization (APCI) - "soft" methods often yielding molecular ions [15]. |
| Key Strength | Excellent chromatographic resolution; highly reproducible, library-searchable EI spectra (e.g., NIST/Wiley); robust and cost-effective operation [15]. | Exceptional sensitivity for polar molecules; broad analyte coverage without need for derivatization; ideal for aqueous biological matrices [2] [15]. |
| Primary Limitation | Requires derivatization for non-volatile/polar metabolites, adding steps, complexity, and potential for error [15]. | Susceptible to matrix effects (ion suppression/enhancement); less reproducible fragmentation patterns; higher operational costs [15] [96]. |
| Identification Power | Strong reliance on extensive, standardized EI spectral libraries for confident identification [15]. | Relies on accurate mass, retention time, MS/MS fragmentation, and comparison with authentic standards; library coverage is improving but less universal [15]. |
| Sample Preparation | Often complex, involving steps for derivatization (e.g., silylation, methoximation) [15]. | Can be simpler (e.g., protein precipitation, dilution) but requires careful management of salts and buffers to prevent ion suppression [15]. |
Coverage and Complementary Nature: No single platform captures the entire metabolome. GC-MS excels for volatile compounds and those made amenable via derivatization, while LC-MS is indispensable for thermally unstable and highly polar metabolites [32]. Studies indicate that using a single platform may identify 100-500 metabolites, but an integrated GC-MS/LC-MS approach can significantly expand coverage, providing a more complete phenotypic snapshot and enhancing the chance of discovering significant biomarkers [32]. Advanced LC-MS configurations, such as dual-column systems combining reversed-phase and hydrophilic interaction chromatography, are emerging to further broaden metabolite coverage within a single run [27].
The Pillars of Reproducibility: Internal Standards and Quality Control Protocols
Reproducibility is engineered into a metabolomics study through rigorous, standardized practices. The Metabolomics Quality Assurance and Quality Control Consortium (mQACC) exemplifies the field's drive to establish best practices [1]. The following workflows and protocols detail how to implement these pillars.
3.1 The Critical Role of Internal Standards An Internal Standard (IS) is a known compound added at a fixed concentration to correct for variability. The calibration is based on the ratio of the analyte response to the IS response, not the absolute response [97]. An IS is most beneficial in multi-step sample preparation protocols (e.g., liquid-liquid extraction, solid-phase extraction) where volumetric losses are common [97].
Selection Criteria:
- Stable Isotope-Labeled (SIL) IS: (e.g., ²H, ¹³C, ¹⁵N) are the gold standard, especially for LC-MS. They have nearly identical chemical and chromatographic properties to the analyte but are distinguishable by mass [96].
- Structural Analogs: Used when SIL-IS are unavailable. Must closely mimic the analyte's extraction efficiency and ionization behavior, but can fail to correct for matrix effects, as demonstrated in Case Study 4 of the search results [96].
- Addition Point: Should be added as early as possible in the workflow (ideally during initial sample aliquoting) to track and correct for all subsequent procedural losses [96].
Monitoring IS Performance: Consistency is key. IS responses should be monitored across a batch. While some variability is normal, investigations are warranted if IS response in unknown samples systematically deviates from that in calibrators, as this may indicate unaccounted matrix effects [96].
3.2 Comprehensive Quality Control Workflow A robust QC strategy employs a suite of samples interleaved within the analytical sequence to monitor and correct for instrumental drift and batch effects [1] [95].
QC Sample Types and Their Purpose:
- Method Blanks: Contain all reagents but no biological matrix. Essential for identifying background contamination from solvents, tubes, or column bleed [95].
- Pooled QC Sample: Created by combining a small aliquot of every study sample. Injected repeatedly throughout the run (e.g., every 6-10 samples) to monitor system stability (retention time drift, signal intensity). Its tight clustering in Principal Component Analysis (PCA) is a key indicator of a stable run [95].
- Calibration Standards: Series of samples with known analyte concentrations, used to construct the quantification curve [97].
- Validation QC Samples: Independently prepared samples at low, medium, and high concentrations, used to assess the method's accuracy and precision during validation and within each batch [95].
Standard Operating Procedures (SOPs): Adherence to detailed, written SOPs for sample collection (e.g., uniform time, container), storage (typically -80°C, minimizing freeze-thaw cycles), metabolite extraction, and instrument tuning is fundamental to minimizing pre-analytical variability [95].
Experimental Protocols for Method Validation
Before applying any method to valuable study samples, it must be rigorously validated. The following table outlines the key parameters to assess, drawing from bioanalytical guidelines applicable to metabolomics [95] [96].
Table 2: Key Method Validation Parameters for Targeted Metabolomics Assays
| Validation Parameter | Experimental Protocol | Acceptance Criteria (Typical) |
|---|---|---|
| Accuracy & Precision | Analyze replicate QC samples (n≥5) at three concentrations (low, mid, high) within a single batch (intra-day) and across different batches/days (inter-day). | Accuracy (% bias): ±15% (±20% at LLOQ). Precision (%CV): ≤15% (≤20% at LLOQ) [95]. |
| Lower Limit of Quantification (LLOQ) | Determine the lowest concentration that can be measured with acceptable accuracy and precision (as defined above). Signal-to-noise ratio is often ≥5:1. | Meets accuracy (±20%) and precision (≤20% CV) criteria [96]. |
| Extraction Recovery & Matrix Effects | Recovery: Compare analyte response from spiked matrix pre-extraction vs. post-extraction. Matrix Effect: Compare analyte response in post-extracted matrix vs. pure solvent. Use SIL-IS to correct for matrix effects. | Recovery should be consistent and reproducible. Matrix factor (IS-normalized) should be close to 1.0 [96]. |
| Carryover | Inject a blank sample immediately after a high-concentration calibration standard or QC. | Analyte response in the blank should be ≤20% of LLOQ response. |
| Stability | Evaluate analyte stability under conditions encountered (bench-top, in-autosampler, freeze-thaw cycles). | Concentration change should be within ±15% of nominal. |
Decision Framework and Concluding Recommendations
Selecting the optimal platform and ensuring reproducibility requires a strategic approach. This framework guides the decision-making process.
Table 3: Decision Framework for Platform Selection in NP Metabolomics
| Primary Decision Driver | Recommended Platform & Rationale | Critical QC Considerations |
|---|---|---|
| Analyte Chemistry (Polarity/Volatility) | Volatile/Fatty Acids/Terpenes: Choose GC-MS for superior separation and library ID [15]. Polar/ Thermolabile (e.g., Flavonoids, Saponins): Choose LC-MS (RP or HILIC) [15]. | GC-MS: Monitor derivatization efficiency batch-to-batch. LC-MS: Use SIL-IS to combat matrix effects; employ heart-cutting 2D-LC for complex mixtures [96] [27]. |
| Study Goal (Targeted vs. Untargeted) | Targeted Quantification of Knowns: The platform best suited for the analyte class (see above). Untargeted Discovery: Consider dual-platform (GC+LC)-MS for maximum coverage [32]. | For untargeted workflows, pooled QC samples are essential for post-acquisition normalization and drift correction across large batches [95]. |
| Available Resources & Expertise | Budget-Limited, High-Throughput Needed: GC-MS often has lower operational costs [15]. Maximizing Sensitivity/Breadth: LC-MS, though costlier, is indispensable for polar biomarkers [3]. | Factor in the cost and availability of appropriate SIL internal standards, which are critical for high-quality quantification but can be expensive [96]. |
Conclusion: Reproducible natural product metabolomics is not achieved by a single instrument but is built through informed platform selection and meticulous attention to quality assurance. GC-MS and LC-MS are complementary pillars of this analytical enterprise. The consistent application of validated protocols, anchored by appropriate internal standards and a comprehensive QC sample strategy, transforms raw data into reliable, biologically meaningful insights. For the most comprehensive profiling, integrating both platforms, governed by these rigorous QC principles, represents the current gold standard in the field [32].
The Scientist's Toolkit: Essential Reagents & Materials for QC
Table 4: Key Research Reagent Solutions for Reproducible Metabolomics
| Item | Function & Importance | Application Notes |
|---|---|---|
| Stable Isotope-Labeled Internal Standards (SIL-IS) | Correct for matrix effects and procedural losses; essential for accurate quantification in LC-MS [96]. | Ideally one per analyte class. Should be added at the very first step of sample processing [1]. |
| Derivatization Reagents (for GC-MS) | Increase volatility and thermal stability of non-volatile metabolites (e.g., sugars, organic acids). Common reagents: MSTFA (N-Methyl-N-(trimethylsilyl)trifluoroacetamide), methoxyamine hydrochloride [15]. | Derivatization is a critical source of variability; conditions (time, temperature) must be strictly controlled and documented in SOPs. |
| Certified Reference Materials & Calibrators | Provide a known concentration to construct the calibration curve, enabling absolute quantification [95]. | Should be prepared in a matrix matching the study samples as closely as possible (e.g., synthetic urine, stripped plasma). |
| Pooled QC Matrix | A homogenized pool of study samples used to monitor instrumental performance and correct for signal drift throughout the run [95]. | Must be prepared in a single, large batch, aliquoted, and stored to ensure consistency across the entire study. |
| Quality Control Samples (Low, Med, High) | Independently prepared samples at known concentrations to verify the method's accuracy and precision within each analytical batch [95]. | Prepared from a separate stock solution than the calibrators to ensure independence. |
| Injection Solvent & Mobile Phase Additives | Must be LC-MS grade to minimize background noise and contamination. Common additives: Formic acid, ammonium acetate/ formate [15]. | Consistency in mobile phase preparation (pH, buffer concentration) is critical for reproducible retention times in LC-MS. |
Strategic Platform Selection and the Case for Integration
Within the ongoing thesis on optimal analytical platforms for natural product metabolomics, this guide provides a direct technical comparison of Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS). The choice between these two cornerstone technologies fundamentally shapes the experimental design, data output, and biological conclusions of a metabolomics study [15] [20].
LC-MS is characterized by its exceptional sensitivity, particularly for polar and thermally labile compounds, and its broad, flexible metabolite coverage, making it the dominant platform for modern biomarker discovery and systems biology [98] [12]. GC-MS, often termed the "gold standard," offers superior reproducibility, robust compound identification via extensive spectral libraries, and excellent chromatographic resolution, especially for volatile and semi-volatile metabolites [20] [4].
The most effective strategy for comprehensive natural product research is not to select a single "best" platform but to deploy LC-MS and GC-MS as complementary tools. This integrated approach maximizes coverage of the chemically diverse metabolome, from primary metabolites and lipids to volatile organic compounds and xenobiotics [99] [12].
Table 1: Core Technical Profile and Complementary Roles
| Comparison Dimension | LC-MS | GC-MS | Complementary Role in Natural Product Research |
|---|---|---|---|
| Optimal Analyte Profile | Polar, ionic, thermolabile; wide MW range (small molecules to peptides) [15] [12]. | Volatile, thermally stable; typically <500-650 Da [15] [4]. | LC-MS: Phenolics, alkaloids, most lipids, peptides. GC-MS: Volatiles, organic acids, sugars, fatty acids, sterols [14] [20]. |
| Primary Ionization | Electrospray Ionization (ESI) - "soft," preserves molecular ion [14] [20]. | Electron Impact (EI) - "hard," generates reproducible fragment patterns [14] [4]. | EI provides universal, searchable spectra; ESI enables detection of intact, labile molecules. |
| Typical Workflow | Minimal derivatization; direct analysis of extracts [15] [61]. | Often requires chemical derivatization (e.g., silylation) for volatility [15] [4]. | Enables analysis of otherwise inaccessible compound classes (e.g., sugars, amino acids by GC-MS). |
| Key Strength | Unparalleled sensitivity and broad, untargeted coverage [14] [98]. | Excellent reproducibility, resolution, and confident identification [99] [4]. | Combined, they deliver both depth (sensitivity) and confidence (identification) across the metabolome. |
Quantitative Performance Comparison
The performance differential between LC-MS and GC-MS is quantifiable across sensitivity, reproducibility, and coverage metrics. The following tables consolidate experimental data from comparative studies.
Table 2: Quantitative Comparison of Sensitivity and Reproducibility
| Parameter | LC-MS Performance | GC-MS Performance | Supporting Experimental Data |
|---|---|---|---|
| Theoretical Sensitivity | ~10⁻¹⁵ mol (femtomole) [14]. | ~10⁻¹² mol (picomole) [14]. | Platform-level theoretical limit comparison. |
| Comparative LOD in Environmental Analysis | Lower detection limits for a panel of pharmaceuticals and personal care products (PPCPs) in water [100]. | Higher detection limits for the same PPCP panel compared to LC-TOF-MS [100]. | Direct method comparison for identical compounds and samples [100]. |
| Chromatographic Reproducibility | Retention time stability can be affected by mobile phase composition and column aging. | Highly reproducible retention times and retention indices (RI) due to stable gas-phase conditions [4]. | GC-MS databases commonly use RI as a second identification parameter [4]. |
| Spectral Reproducibility | Fragmentation (MS/MS) patterns can vary with instrument and collision energy. | Electron Ionization (EI) mass spectra are highly reproducible across instruments and over time, enabling universal libraries [15] [4]. | The NIST library contains >240,000 reproducible EI spectra [4]. |
| Peak Capacity (Complex Samples) | High, especially with UHPLC and multidimensional separations (e.g., HILIC + RPLC) [12] [20]. | High for 1D-GC; significantly increased with comprehensive 2D-GC (GC×GC). | In serum analysis, GC×GC-TOFMS detected ~3x as many peaks as 1D GC-MS at SNR≥50 [99]. |
Table 3: Quantitative Comparison of Metabolite Coverage and Identification
| Parameter | LC-MS Performance | GC-MS Performance | Supporting Experimental Data |
|---|---|---|---|
| Untargeted Peak Detection | Capable of detecting thousands of features in a single run from complex extracts [98] [20]. | Fewer features typically detected compared to LC-MS, but with high confidence. | In a serum study, GC×GC-MS identified 3x the number of metabolites (Rsim≥600) vs. 1D GC-MS [99]. |
| Confident Compound Identification | Relies on accurate mass, MS/MS, and retention time; library spectra are less universal [15]. | Strong identification via mass spectrum and retention index matching against large, standardized libraries [4]. | NIST GC-MS library: >240,000 compounds. NIST LC-MS/MS library: ~8,171 compounds [4]. |
| Biomarker Discovery Yield | High yield due to broad coverage; dominant platform in clinical metabolomics [98]. | Robust for its chemical domain; can reveal unique biomarkers. | In a neurodegenerative disease study, 34 significant metabolites were found by GC×GC-MS vs. 23 by GC-MS, with 9 overlapping [99]. |
| Coverage of Chemical Space | Exceptional for polar, ionic, and high molecular weight compounds (e.g., lipids, flavonoids) [14] [12]. | Excellent for volatile, non-polar, and thermally stable small molecules (e.g., organic acids, sugars after derivatization) [20]. | Over 70% of metabolites are non-volatile, favoring LC-MS as the primary tool [12]. |
Detailed Experimental Methodologies
The quantitative differences summarized above arise from distinct foundational methodologies. Below are detailed protocols representative of each platform.
This protocol highlights the derivatization required for GC-MS and the use of pooled quality control (QC) samples to monitor reproducibility.
Sample Preparation & Extraction:
- Add 100 µL of serum to 1 mL of ice-cold methanol/chloroform (3:1 v/v) containing internal standards (e.g., heptadecanoic acid, norleucine).
- Vortex and centrifuge (15 min, 18,000 rcf, 4°C). Transfer supernatant and dry under a gentle N₂ stream.
Chemical Derivatization (Critical for Volatility):
- Methoximation: Add 50 µL of methoxyamine in pyridine (20 mg/mL). Shake (90 min, 30°C).
- Silylation: Add 50 µL of N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) with 1% TMCS. Shake (60 min, 70°C).
Instrumental Analysis (GC-TOFMS):
- Column: 30-60m DB-5ms (or equivalent) capillary column.
- Temperature Program: 60°C hold 1 min, ramp at 5°C/min to 300°C, hold 12 min.
- Ionization: Electron Impact (EI) at 70 eV.
- QC Strategy: Inject a pooled QC sample after every 9-10 biological samples to monitor system stability and enable data correction.
Data Processing:
- Use deconvolution software (e.g., AMDIS, ChromaTOF) to separate co-eluting peaks.
- Align peaks across samples and identify compounds by matching mass spectra and retention indices to libraries (e.g., NIST, FiehnLib).
This protocol emphasizes the "soft" ionization and broad separation strategies central to LC-MS.
Sample Preparation & Extraction:
- Precipitate proteins by adding cold methanol or acetonitrile (typically 3:1 or 4:1 solvent-to-sample ratio) to plasma/serum.
- Vortex, centrifuge, and collect the supernatant. Often includes a lipid removal step for polar metabolomics.
Liquid Chromatography Separation:
- For Polar Metabolites (HILIC): Use a hydrophilic column (e.g., silica, amide). Mobile phase: (A) water with buffer, (B) acetonitrile. Start high %B.
- For Lipids & Mid-Polar Metabolites (RPLC): Use a hydrophobic column (e.g., C18). Mobile phase: (A) water with acid, (B) methanol or acetonitrile. Start low %B.
- Modern Platform: Ultra-High Performance LC (UHPLC) with sub-2µm particles for high resolution and speed.
Mass Spectrometry Analysis (Q-TOF or Orbitrap):
- Ionization: Electrospray Ionization (ESI) in positive and/or negative mode.
- Data Acquisition: Full-scan high-resolution MS (for accurate mass) combined with data-dependent MS/MS (for fragmentation spectra).
- QC Strategy: Use pooled QC samples and solvent blanks. Acquire QC frequently to assess instrument performance.
Data Processing & Identification:
- Process raw data (peak picking, alignment, integration) using software (e.g., MS-DIAL, XCMS).
- Annotate metabolites using accurate mass (±5 ppm), isotopic pattern, and MS/MS spectra matched to public (e.g., MassBank, GNPS) or in-house libraries.
Visualizing the Complementary Workflow
The strategic integration of LC-MS and GC-MS creates a powerful, comprehensive metabolomics pipeline.
LC-MS and GC-MS Complementary Analysis Workflow
Platform Selection Decision Tree for Natural Product Research
The Scientist's Toolkit: Essential Reagents and Materials
Successful implementation of the protocols above requires specific reagents and materials. This toolkit categorizes essentials by platform and function.
Table 4: Research Reagent Solutions for LC-MS and GC-MS Metabolomics
| Category | Item | Primary Function | Platform |
|---|---|---|---|
| Sample Preparation | Methanol, Acetonitrile, Chloroform, Isopropanol | Metabolite extraction and protein precipitation from biological matrices [99] [4]. | Both |
| Internal Standards (e.g., Heptadecanoic acid, Norleucine, Isotopically-labeled analogs) | Monitors extraction efficiency, corrects for instrument variability, enables quantification [99] [4]. | Both | |
| Derivatization (GC-MS) | Methoxyamine hydrochloride | Protects carbonyl groups (aldehydes, ketones) by forming methoximes during derivatization [99] [4]. | GC-MS |
| N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) with 1% TMCS | Replaces active hydrogens (-OH, -COOH, -NH) with trimethylsilyl groups, rendering metabolites volatile and stable for GC [99] [4]. | GC-MS | |
| Pyridine | Solvent for methoximation and silylation reactions [99]. | GC-MS | |
| Chromatography | GC Columns: DB-5ms (non-polar), DB-17ms (mid-polar) | Separate metabolites based on volatility and interaction with stationary phase. A primary (DB-5) and secondary (DB-17) column are used for GC×GC [99]. | GC-MS |
| LC Columns: C18 (Reverse Phase), HILIC (Hydrophilic Interaction) | Separate metabolites based on polarity. C18 for lipids/mid-polar compounds; HILIC for polar metabolites [12] [20]. | LC-MS | |
| Ultra-pure Helium Gas | Inert carrier gas for moving vaporized samples through the GC column [99] [61]. | GC-MS | |
| LC-MS Grade Solvents with Additives (e.g., Water, MeOH, ACN, Formic Acid, Ammonium Acetate) | Mobile phase for LC separation. Additives (acids, buffers) modulate pH and improve ionization efficiency [101] [100]. | LC-MS | |
| Calibration & QC | Alkane Retention Index Standard (C10-C40) | Allows calculation of retention indices (RI) for metabolite identification in GC-MS [99] [4]. | GC-MS |
| FlexMix or Calmix Calibration Solution | Provides standard ions for mass accuracy calibration of the MS system [101]. | LC-MS (Orbitrap) | |
| Pooled Quality Control (QC) Sample | A homogeneous sample injected repeatedly throughout the batch to monitor and correct for instrumental drift [99] [98]. | Both |
In natural product metabolomics research, the choice between Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass Spectrometry (GC-MS) is foundational and analyte-dependent [18] [14]. No single platform can capture the entire chemical diversity of plant extracts, which contain hundreds to thousands of metabolites spanning a wide range of polarities, volatilities, and molecular weights [18]. The classical approach of bioassay-guided fractionation risks losing synergistic biological information present in whole extracts, creating a need for comprehensive analytical profiling [18]. This guide provides an objective, data-driven comparison to enable researchers to select the optimal platform based on the chemical classes of interest—volatiles, lipids, and polar metabolites—within the broader methodological thesis of LC-MS versus GC-MS.
Platform Comparison: Performance Specifications and Capabilities
The core specifications of LC-MS and GC-MS differ significantly, leading to complementary strengths. The following tables summarize their key characteristics, supported by experimental data.
Table 1: Core Platform Characteristics and Performance Metrics [99] [14] [102]
| Feature | GC-MS (including GC×GC-MS) | LC-MS | Implication for Natural Product Research |
|---|---|---|---|
| Ionization Source | Electron Impact (EI) [14] | Electrospray (ESI), Atmospheric Pressure Chemical Ionization (APCI) [14] | EI produces reproducible, fragment-rich spectra ideal for library matching. ESI/APCI better preserves molecular ions for mass accuracy-based identification. |
| Typical Sensitivity | ~10⁻¹² mol [14] | ~10⁻¹⁵ mol [14] | LC-MS generally offers higher sensitivity, crucial for detecting low-abundance secondary metabolites. |
| Chromatographic Separation | Gas phase (inert carrier); high-resolution with long or multi-dimensional columns [99] [14]. | Liquid phase (solvent gradient); versatile with multiple column chemistries (C18, HILIC, etc.). | GC excels for volatiles. LC offers flexible separation for non-volatile and thermally labile compounds like many glycosides. |
| Sample Preparation | Often requires chemical derivatization for non-volatile polar metabolites (e.g., silylation) [99]. | Typically minimal; direct injection of extracts after filtration or dilution [18]. | GC-MS prep is more time-consuming and can introduce artifacts. LC-MS offers faster, simpler workflows. |
| Metabolite Identification | Relies on universal, reproducible EI spectral libraries (e.g., NIST) [99] [103]. | Relies on accurate mass, MS/MS fragmentation, and compound-specific libraries [103]. | GC-MS IDs are highly reliable. LC-MS identification is powerful but can be more ambiguous without authentic standards. |
Table 2: Analyte-Specific Coverage and Experimental Performance Data
| Analyte Class | Recommended Platform | Key Experimental Findings & Coverage | Supporting Evidence |
|---|---|---|---|
| Volatile Organic Compounds (VOCs) | GC-MS (with HS-SPME or ITEX) [104] [102] | Ideal for aldehydes, alcohols, ketones, terpenes, esters. A study identified 35 VOCs (aldehydes, alcohols, ketones, furans) in fruit kernels [102]. Specialized extraction (e.g., in-tube extraction) enables exhaustive profiling of oxidized lipid-derived volatiles like 1-octen-3-ol [104]. | [104] [102] |
| Lipids (Non-volatile) | LC-MS (especially RP-LC) [58] [14] | Superior for intact phospholipids, triglycerides, glycolipids. GC-MS is unsuitable for large, non-volatile lipids but can analyze fatty acid methyl esters (FAMEs) after hydrolysis/derivatization. | [58] [14] |
| Polar Primary Metabolites | Either Platform (with derivation for GC) | GC×GC-MS detected ~3x more peaks and identified ~3x more metabolites from human serum than 1D GC-MS at high confidence (SNR≥50, Rₛᵢₘ≥600) [99]. LC-MS (e.g., with HILIC) can analyze these (sugars, organic acids, amino acids) without derivation. | [99] |
| Secondary Metabolites | LC-MS (primary), GC-MS for some | LC-MS is preferred for flavonoids, anthocyanins, alkaloids, saponins due to their polarity and thermal lability [14]. GC-MS can analyze some terpenoids and phenolics after derivation. | [18] [14] |
Detailed Experimental Protocols from Key Studies
Protocol 1: Comprehensive GC×GC-MS vs. GC-MS Analysis for Serum Metabolomics [99]
- Sample: 100 µL of human serum.
- Extraction: Add to 1 mL ice-cold methanol/chloroform (3:1 v/v) with internal standards. Vortex, centrifuge (18,000 rcf, 15 min, 4°C). Dry supernatant under N₂.
- Derivatization: Two-step method. 1) Add 50 µL methoxyamine in pyridine (20 mg/mL), shake (90 min, 30°C). 2) Add 50 µL MSTFA with 1% TMCS, shake (60 min, 70°C).
- GC-MS Analysis: Column: DB-5ms UI (60 m x 0.25 mm, 0.25 µm). Splitless injection. Oven: 60°C (1 min) to 300°C at 5°C/min, hold 12 min.
- GC×GC-MS Analysis: 1D column: same as GC-MS. 2D column: DB-17ms (1 m x 0.25 mm, 0.25 µm). Modulator period: 2.5 s. Secondary oven offset: +10°C.
- MS Detection (Both): LECO Pegasus TOF-MS. EI source (-70 eV). Mass range: m/z 45-1000.
- Data Analysis: Use ChromaTOF for peak picking, then MetPP software for peak alignment and RI matching (p ≤ 0.001) [99].
Protocol 2: HS-SPME-GC-MS Profiling of Volatile and Non-Volatile Metabolites in Plant Kernels [102]
- Sample Preparation: Kernels are lyophilized and ground. For VOCs: Use headspace solid-phase microextraction (HS-SPME).
- HS-SPME: Incubate sample in vial. Expose SPME fiber to headspace to adsorb volatiles.
- GC-MS Analysis: Inject thermally desorbed volatiles into GC. Identify compounds using spectral libraries.
- For Polar/Lipid Metabolites: Extract ground kernel material with suitable solvent (e.g., methanol/water/chloroform). Derivatize (methoxyamination and silylation) for GC-MS analysis of polar metabolites [102].
Protocol 3: Exhaustive Profiling of Oxidized Lipid-Derived Volatiles via ITEX-GC-MS [104]
- Oxidation: Oxidize phosphatidylcholine standards or plasma with radical initiator AAPH (0.5 M in PBS) at 37°C for 4 hours with agitation.
- Extraction: Use in-tube extraction (ITEX). Transfer 100 µL oxidized sample to vial with baked NaCl. Agitate at 500 rpm, 50°C. Perform 10 extraction strokes with a Tenax TA-packed syringe.
- GC-MS Analysis: Column: Omegawax 250 (30 m x 0.25 mm, 0.25 µm). Oven: 35°C (2 min) to 230°C at 4°C/min, hold 15 min. MS in full-scan mode (m/z 35-300).
- Key Advantage: ITEX allows exhaustive trapping of diverse volatiles (alcohols, ketones, furanones) beyond common aldehydes [104].
Workflow and Decision-Making Diagrams
Platform Selection Workflow for Natural Product Analysis
Comprehensive GCxGC-TOFMS Metabolomics Workflow [99]
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Materials for Metabolomics Sample Preparation
| Item | Function | Typical Application/Note |
|---|---|---|
| Methanol, Chloroform, Water (LC-MS grade) | Primary extraction solvents for the "Folch" or "Bligh & Dyer" methods [18] [105]. | Used in specific ratios (e.g., 2:1:0.8 v/v/v) to simultaneously extract polar metabolites and lipids [18] [105]. |
| Methyl tert-butyl ether (MTBE) | Alternative, safer extraction solvent to chloroform for liquid-liquid partitioning [18]. | Particularly used for lipidomics and combined metabolite/lipid extraction from diverse samples [18]. |
| Methoxyamine hydrochloride | First derivatization reagent; protects carbonyl groups by forming methoximes [99]. | Reduces formation of multiple sugar anomers and decarboxylation products during GC-MS analysis [99]. |
| N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) | Silylation reagent; replaces active hydrogens with trimethylsilyl groups, increasing volatility [99]. | Critical for GC-MS analysis of polar metabolites like sugars, organic acids, and amino acids [99]. |
| Heptadecanoic Acid, Norleucine | Internal standards (IS) for quantification [99]. | Added at known concentration before extraction to correct for losses during preparation and instrument variability [99]. |
| 2,2'-Azobis(2-amidinopropane) dihydrochloride (AAPH) | Radical initiator for in vitro oxidation studies [104]. | Used to generate oxidized lipid standards and model systems to profile resulting volatile compounds [104]. |
| Tenax TA | Porous polymer adsorbent packed in needles for in-tube extraction (ITEX) [104]. | Exhaustively traps a wide range of volatile compounds from headspace for sensitive GC-MS analysis [104]. |
| Retention Index Standard (Alkanes C10-C40) | Standard mixture for calibrating retention times [99]. | Run periodically to convert retention times to system-independent retention indices for reliable metabolite identification [99]. |
The selection between LC-MS and GC-MS is not a question of superiority but of strategic application. GC-MS, particularly in its comprehensive two-dimensional form (GC×GC-MS), is unparalleled for the discovery and robust identification of volatile metabolites and derivatized polar primary metabolites, offering superior chromatographic resolution and library-dependent identification [99] [104] [102]. LC-MS is the indispensable platform for the direct analysis of non-volatile lipids, thermally labile secondary metabolites, and broad-spectrum untargeted profiling, providing higher sensitivity and flexibility [58] [18] [14].
For a holistic analysis of natural products, a multi-platform approach is strongly recommended. A practical strategy involves: 1) Using LC-MS for an initial untargeted screen to capture a wide metabolite range, 2) Employing GC-MS for targeted, high-confidence identification of volatile and primary metabolites, and 3) Applying specialized techniques like HS-SPME-GC-MS or ITEX-GC-MS for exhaustive volatile profiling [104] [102]. This integrated methodology aligns with the modern paradigm in natural product metabolomics, which seeks to preserve and understand the complex chemical synergy within biological extracts [18].
The comprehensive analysis of small-molecule metabolites, or metabolomics, has emerged as a pivotal tool for understanding the complex chemistry and biological activity of natural products (NPs) [41]. Within the broader thesis context of LC-MS versus GC-MS for natural product metabolomics, the fundamental choice between untargeted and targeted analytical strategies critically shapes research outcomes [106] [32]. Untargeted metabolomics provides a global, hypothesis-generating profile of a sample's metabolome, capturing both known and unknown compounds. In contrast, targeted metabolomics delivers precise, quantitative data on a predefined set of metabolites, validating specific biochemical hypotheses [106] [107]. The inherent chemical diversity of NPs—spanning polar, semi-polar, and non-polar compounds—makes the selection of the analytical platform (LC-MS or GC-MS) inseparable from the choice of metabolomics approach [41] [32]. This guide objectively compares the performance of these paired strategies, providing researchers and drug development professionals with a framework to align their analytical methodology with their scientific objectives in NP research.
Comparative Performance of Untargeted and Targeted Metabolomics
The core distinctions between untargeted and targeted metabolomics are defined by their scope, objectives, and performance metrics. The following tables synthesize key quantitative and qualitative differences to guide platform selection.
Table 1: Core Conceptual and Performance Comparison of Untargeted vs. Targeted Metabolomics
| Feature | Untargeted Metabolomics | Targeted Metabolomics |
|---|---|---|
| Primary Goal | Discovery, hypothesis generation, and comprehensive metabolic profiling [106] [108]. | Hypothesis validation and precise quantification of known metabolites [106] [107]. |
| Analytical Scope | Broad; aims to detect all measurable metabolites (100s to 1000s), including unknowns [106] [32]. | Narrow; focuses on a predefined set of characterized analytes (typically ~20, but up to 100s in semi-targeted) [106] [109] [108]. |
| Quantification | Relative (semi-quantitative); compares metabolite abundance between samples [106] [107]. | Absolute; uses calibration curves and internal standards for precise concentration measurements [106] [107]. |
| Sensitivity & Precision | Variable sensitivity; lower precision due to relative quantification and lack of specific optimization [106] [107]. | High sensitivity and precision for target analytes, optimized via specific protocols [106] [107]. |
| Data Complexity | High; generates large, complex datasets requiring advanced bioinformatics for processing and analysis [106] [32]. | Lower; yields focused data that is more straightforward to analyze and interpret [107]. |
| Ideal Application in NP Research | Biomarker discovery, quality control via metabolic fingerprinting, and uncovering novel bioactive compounds [41]. | Validating marker compounds, pharmacokinetic studies, and batch-to-batch consistency testing of known actives [41]. |
Table 2: Platform-Driven Performance: LC-MS vs. GC-MS in Natural Product Metabolomics
| Performance Criteria | LC-MS (Untargeted/Targeted) | GC-MS (Untargeted/Targeted) | Integrated LC-MS + GC-MS [32] |
|---|---|---|---|
| Optimal Compound Coverage | Semi-polar to polar, non-volatile, and thermally labile molecules (e.g., flavonoids, saponins, most alkaloids) [32] [3]. | Volatile, thermally stable, and derivatized polar compounds (e.g., essential oils, organic acids, sugars after derivatization) [32] [3]. | Most comprehensive coverage. Combines strengths of both to profile a vastly broader range of metabolite classes [32]. |
| Typical Metabolite ID Range (Untargeted) | Up to ~500 metabolites per analysis [32]. | Up to ~100 metabolites per analysis [32]. | ~353+ metabolites identified in a reference plasma study, demonstrating enhanced coverage [32]. |
| Sample Preparation | Generally simpler; often involves dilution or simple solvent extraction [78] [3]. | Often requires chemical derivatization for non-volatile compounds, adding steps and complexity [32] [78]. | Most complex, requiring protocols compatible with both platforms [32]. |
| Throughput & Run Time | Shorter run times; faster due to minimal preparation and no derivatization [78]. | Longer run times; slower due to derivatization steps and typical GC column gradients [78]. | Lowest throughput; essentially doubles analytical time and cost [32]. |
| Operational Cost | Higher initial instrument cost; higher cost of solvents and maintenance [3]. | Lower initial instrument and operational cost [61] [3]. | Highest cost, combining expenses of both platforms [32]. |
| Key Application in NP Analysis | Profiling of most secondary metabolites (e.g., ginsenosides, berberine, curcuminoids) [41] [3]. | Analysis of volatiles, fatty acids, and primary metabolites (sugars, amino acids) [41] [32]. | Creating complete metabolic fingerprints for superior quality control and biomarker discovery [41] [32]. |
Detailed Experimental Protocols for Key Applications
The selection of a protocol is contingent upon the chosen analytical strategy and platform. Below are detailed methodologies for two cornerstone applications in natural product research.
Objective: To obtain a comprehensive, unbiased metabolic profile of a plant extract for quality control or discovery purposes.
Workflow Summary:
- Sample Preparation:
- Extraction: Homogenize 50 mg of freeze-dried plant material with 1 mL of a cold methanol:water (4:1, v/v) mixture. Vortex vigorously for 1 minute and sonicate in an ice bath for 15 minutes.
- Cleaning: Centrifuge at 14,000 x g for 15 minutes at 4°C. Transfer the supernatant to a new vial.
- Splitting: Divide the extract into two equal aliquots. One aliquot is for LC-MS analysis and can be dried and reconstituted in a compatible solvent (e.g., methanol with 0.1% formic acid). The second aliquot is for GC-MS analysis and must be completely dried under a gentle nitrogen stream.
- Derivatization (for GC-MS aliquot): Add 50 µL of methoxyamine hydrochloride (20 mg/mL in pyridine) to the dried aliquot and incubate at 37°C for 90 minutes with shaking. Then, add 50 µL of N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA) and incubate at 37°C for 30 minutes [32].
Data Acquisition:
- LC-MS: Inject sample onto a reversed-phase C18 column. Use a gradient elution with water (A) and acetonitrile (B), both containing 0.1% formic acid. Acquire data in both positive and negative electrospray ionization (ESI) modes on a high-resolution mass spectrometer (e.g., Q-TOF) in data-dependent acquisition (DDA) mode.
- GC-MS: Inject derivatized sample in splitless mode onto a non-polar (e.g., DB-5MS) column. Use helium as carrier gas and a temperature gradient. Acquire data using electron ionization (EI) at 70 eV with full scan monitoring.
Data Processing & Analysis:
- Process raw files using specialized software (e.g., XCMS, MS-DIAL) for peak picking, alignment, and normalization.
- Combine the feature lists from the LC-MS and GC-MS datasets.
- Perform multivariate statistical analysis (e.g., Principal Component Analysis - PCA) on the combined dataset to identify patterns and outliers.
- Annotate significant features by querying accurate mass and fragmentation spectra against public (e.g., GNPS, NIST) and commercial metabolomics libraries.
Objective: To absolutely quantify the concentration of specific, known bioactive markers (e.g., berberine in Coptidis rhizoma) for standardization.
Workflow Summary:
- Preparation of Standards and Samples:
- Calibrants & QCs: Prepare a series of calibration standards and quality control (QC) samples by spiking known amounts of pure reference standards (e.g., berberine) into a surrogate or blank matrix.
- Internal Standard (ISTD): Add a fixed amount of a stable isotope-labeled analog of each target analyte (e.g., berberine-d6) to every sample, calibrant, and QC before extraction to correct for variability.
- Sample Extraction: Weigh 100 mg of ground herbal material. Add ISTD solution and extract with 1 mL of a defined solvent (e.g., 70% methanol) via vortexing and sonication. Centrifuge and dilute the supernatant to within the calibration range.
Data Acquisition (Targeted MS):
- Chromatography: Use optimized, isocratic or short-gradient conditions on an appropriate column (e.g., HILIC for polar compounds) to rapidly separate the target compounds.
- Mass Spectrometry: Operate a triple quadrupole (QQQ) mass spectrometer in Multiple Reaction Monitoring (MRM) mode. For each analyte and ISTD, define a specific precursor ion > product ion transition. Dwell times are optimized for maximum sensitivity and sufficient data points across the peak.
Data Processing & Quantification:
- Integrate the peak areas for each analyte and its corresponding ISTD from the MRM chromatograms.
- Calculate the peak area ratio (Analyte/ISTD) for each calibrant and construct a linear (or weighted) calibration curve.
- Use this curve to calculate the absolute concentration of each target analyte in the unknown samples based on their measured peak area ratios. Apply acceptance criteria (e.g., QC samples within ±15% of nominal value) to validate the run.
Strategic Workflow and Platform Decision Pathways
The following diagrams illustrate the logical workflows for an integrated metabolomics approach and the decision-making process for selecting an analytical platform.
Workflow for Integrated NP Metabolomics
Platform Selection Decision Tree
The Scientist's Toolkit: Essential Research Reagent Solutions
Successful execution of metabolomics studies requires specific, high-quality reagents and materials. This toolkit details essential items for natural product analysis.
Table 3: Essential Research Reagents and Materials for NP Metabolomics
| Item Category | Specific Examples & Specifications | Primary Function in Workflow |
|---|---|---|
| Chromatography Solvents & Additives | LC-MS grade water, acetonitrile, methanol. Formic acid, ammonium acetate or formate [78] [3]. | Forms the mobile phase for LC-MS; high purity minimizes background noise and ion suppression. Additives modify pH and improve ionization. |
| Derivatization Reagents (for GC-MS) | Methoxyamine hydrochloride, N-Methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA), N,O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA) [32] [78]. | Chemically modifies polar, non-volatile metabolites (sugars, organic acids) to volatile, thermally stable trimethylsilyl derivatives for GC-MS analysis. |
| Isotopically Labeled Internal Standards | Stable isotope-labeled analogs of target metabolites (e.g., Berberine-d6, Ginsenoside Rg1-d3). Certified reference materials from suppliers like Cerilliant [78]. | Essential for targeted quantification. Corrects for analyte loss during sample preparation and matrix-induced ionization suppression/enhancement in MS. |
| Reference Standards & Libraries | Pure, certified chemical standards of known NP metabolites (e.g., curcumin, baicalin). Commercial (NIST, Wiley) and public (GNPS, MassBank) spectral libraries [41] [3]. | Used for calibrating targeted assays and for annotating/identifying metabolites in untargeted workflows by matching retention time and mass spectra. |
| Solid-Phase Extraction (SPE) Sorbents | Mixed-mode (cation/anion exchange + reversed-phase), C18, polymer-based cartridges [78]. | Selectively cleans complex NP extracts, removes salts and interfering matrix components, and pre-concentrates analytes to improve sensitivity. |
| Enzymes for Hydrolysis | β-Glucuronidase/Sulfatase (e.g., from Helix pomatia or recombinant) [78]. | Deconjugates phase II metabolites (glucuronides, sulfates) in biological samples back to their parent aglycones for accurate quantification of total content. |
In the field of natural product metabolomics, the comprehensive characterization of complex chemical mixtures is a fundamental challenge. No single analytical platform can capture the full physicochemical diversity of metabolites, which range from volatile terpenes and polar organic acids to high-molecular-weight, non-volatile glycosides and lipids [110]. This limitation forces a critical choice: Gas Chromatography-Mass Spectrometry (GC-MS) or Liquid Chromatography-Mass Spectrometry (LC-MS) [15].
Traditionally viewed as competitors, these techniques are in fact powerfully complementary. GC-MS excels in the analysis of volatile and thermally stable compounds, offering unparalleled chromatographic resolution and access to robust, universal spectral libraries [15] [14]. LC-MS, in contrast, is indispensable for polar, ionic, and thermally labile molecules, operating at ambient temperatures and providing exceptional sensitivity for bioanalysis [2] [3]. The central thesis of this guide is that for untargeted profiling—where the goal is to capture as many metabolites as possible without a priori knowledge—the synergistic integration of GC-MS and LC-MS is not merely beneficial but essential for achieving a holistic view of the metabolome [111] [110].
This comparison guide objectively evaluates the performance of both platforms, supported by experimental data, to provide researchers and drug development professionals with a framework for designing robust, comprehensive metabolomics studies.
Foundational Comparison: Principles and Performance
The operational divergence between GC-MS and LC-MS stems from their fundamental separation and ionization mechanisms, which directly dictate their application scope and performance characteristics.
GC-MS relies on gas-phase separation. Analytes must be volatile or be chemically derivatized to become volatile, and they must withstand the high temperatures required for vaporization and chromatography. Separation occurs in a heated capillary column based on analyte boiling points and interactions with the stationary phase, typically using an inert carrier gas like helium [15] [14]. Ionization is most commonly achieved via Electron Ionization (EI), a "hard" method that generates extensive, reproducible fragment patterns ideal for library matching against established databases like NIST and Wiley [15] [112].
LC-MS employs liquid-phase separation at ambient or controlled temperatures, making it suitable for thermolabile compounds. Separation occurs in a column packed with solid particles, based on differential partitioning between a liquid mobile phase and the stationary phase [3]. Ionization is "soft," typically using Electrospray Ionization (ESI) or Atmospheric Pressure Chemical Ionization (APCI), which often produces intact molecular ions with minimal fragmentation, preserving information about the parent molecule [15] [14].
The following table summarizes the core technical and performance differences between the two platforms.
Table 1: Foundational Comparison of GC-MS and LC-MS Platforms
| Aspect | GC-MS | LC-MS |
|---|---|---|
| Separation Principle | Gas-phase partitioning; high temperature required [15]. | Liquid-phase partitioning; ambient/moderate temperature [3]. |
| Ideal Analyte Properties | Volatile, thermally stable, typically <500 Da [15]. | Polar, ionic, thermally labile; broad mass range (small molecules to proteins) [2] [3]. |
| Typical Ionization | Electron Ionization (EI) – hard, reproducible fragmentation [15] [14]. | Electrospray (ESI) / APCI – soft, often yields molecular ion [15] [14]. |
| Key Strength | Excellent resolution for structural isomers; universal, reproducible spectral libraries [15] [110]. | Extremely broad analyte coverage; superior sensitivity for many polar biomolecules [2] [3]. |
| Major Limitation | Often requires derivatization for non-volatile compounds [15] [110]. | Susceptible to matrix effects; lacks universal spectral library [78] [15]. |
| Common Applications | Essential oils, VOCs, fatty acids, organic acids (after derivatization), environmental pollutants [15] [3]. | Pharmaceuticals, peptides, lipids, polar natural products (e.g., flavonoids, glycosides), clinical metabolomics [2] [3]. |
Experimental Performance Data: A Direct Comparison
A comparative study analyzing five benzodiazepines in urine provides concrete, quantitative data on the performance of LC-MS/MS versus GC-MS [78]. Both methods were validated for linearity, precision, and accuracy around a 100 ng/mL decision point.
Table 2: Experimental Performance Comparison for Benzodiazepine Analysis [78]
| Parameter | GC-MS Performance | LC-MS/MS Performance | Comparative Insight |
|---|---|---|---|
| Average Accuracy | 99.7% - 107.3% | 99.7% - 107.3% | Both techniques delivered equivalent and excellent accuracy. |
| Average Precision (%CV) | <9% | <9% | Both techniques showed comparable and acceptable precision. |
| Sample Preparation | Complex: Enzymatic hydrolysis, SPE, derivatization with MTBSTFA (85 min total) [78]. | Simplified: Enzymatic hydrolysis, SPE (no derivatization) [78]. | LC-MS/MS workflow is significantly faster and less complex. |
| Run Time | Longer chromatographic cycle. | Shorter chromatographic cycle [78]. | LC-MS/MS offers higher throughput. |
| Matrix Effects | Not prominently reported. | Observed for all analytes; controlled by deuterated internal standards [78]. | LC-MS/MS is more susceptible to ionization suppression/enhancement, requiring robust internal standardization. |
| Identification Specificity | Relies on retention time and EI spectrum matching. | Relies on retention time and multiple reaction monitoring (MRM) transitions. | Both are highly specific when properly configured. |
Key Experimental Insight: The study concluded that while both methods produced analytically equivalent results, the ease of sample preparation, broader analyte coverage, and shorter run time made LC-MS/MS a more expedient confirmation technology in a high-throughput setting [78]. This highlights a practical advantage of LC-MS, though the inherent requirement for GC-MS to analyze certain volatile compounds remains unchanged.
The Case for Integration: Coverage and Workflow
The fundamental rationale for integration is complementary metabolite coverage. GC-MS and LC-MS access different, often non-overlapping, regions of the metabolome due to their inherent physicochemical requirements [110].
Integrated Platform Coverage of the Metabolome
An integrated untargeted workflow leverages these complementary strengths. The typical process involves parallel sample processing streams that converge during data analysis [112].
Integrated GC-MS and LC-MS Untargeted Profiling Workflow
Instrumentation and Technical Specifications
Modern advances in both platforms have expanded their capabilities. High-resolution accurate mass (HRAM) analyzers, like Orbitrap and Q-TOF systems, are now common in LC-MS, providing exact mass measurements for confident formula assignment [2] [113]. GC-MS benefits from two-dimensional chromatography (GC×GC) for separating extremely complex volatile mixtures [14].
Table 3: Specifications of Representative High-Resolution LC-MS Systems (Orbitrap-Based) [113]
| Model | Resolving Power (@ m/z 200) | Mass Accuracy | Scan Speed | Ideal Applications |
|---|---|---|---|---|
| Orbitrap Exploris 120 | 120,000 | <1 ppm (with EASY-IC) | 22 Hz | Food/Env. safety, targeted/semi-targeted metabolomics [113]. |
| Orbitrap Exploris 240 | 240,000 | <1 ppm (with EASY-IC) | Up to 22 Hz | Untargeted metabolomics, lipidomics, forensic toxicology [113]. |
| Q Exactive Plus | 140,000 (up to 280,000) | <1 ppm (internal cal.) | 12 Hz | Metabolomics, lipidomics, biopharma development [113]. |
Experimental Protocols for Comparative Analysis
The following protocols are adapted from the benzodiazepine comparison study, illustrating the distinct sample preparation requirements for each platform [78].
Protocol A: GC-MS Analysis for Benzodiazepines
- Sample Preparation: To 1 mL of urine, add 0.100 mL of deuterated internal standard (ISTD), 2 mL of 0.1 M sodium acetate buffer (pH 4.75), and 0.050 mL of β-glucuronidase.
- Hydrolysis: Vortex, then incubate for 60 minutes at 55°C to hydrolyze glucuronide conjugates.
- Solid-Phase Extraction (SPE): After cooling and centrifugation, load supernatant onto a CEREX CLIN II SPE cartridge. Wash sequentially with carbonate buffer (pH 9), water-acetonitrile (80:20), and water. Dry cartridge for 15 minutes.
- Elution & Evaporation: Elute analytes with 1 mL of methylene chloride-methanol-ammonium hydroxide (85:10:2). Evaporate eluent to dryness at 55°C.
- Derivatization: Add 0.050 mL each of ethyl acetate and MTBSTFA (with 1% MTBDMCS) to the dried extract. Incubate for 20 minutes at 65°C.
- GC-MS Analysis: Inject 0.5 µL onto an Agilent HP-ULTRA 1 column. Use helium carrier gas and a temperature gradient. Detect via mass spectrometry with electron ionization (EI) [78].
Protocol B: LC-MS/MS Analysis for Benzodiazepines
- Sample Preparation/Hydrolysis: Identical first steps to Protocol A (through enzymatic hydrolysis).
- Solid-Phase Extraction (SPE): Use a different SPE cartridge (Clean Screen XCEL I). Follow a similar wash procedure but with buffers optimized for LC-MS compatibility.
- Elution & Reconstitution: Elute with an appropriate solvent (e.g., methanol with ammonium hydroxide). Evaporate and reconstitute in LC mobile phase. Note: No derivatization step is required.
- LC-MS/MS Analysis: Inject onto a reversed-phase LC column. Use a gradient of water and acetonitrile, both with 0.1% formic acid. Analyze via tandem mass spectrometry using electrospray ionization (ESI) in positive mode and Multiple Reaction Monitoring (MRM) [78].
The Scientist's Toolkit: Essential Reagents and Materials
Table 4: Key Reagents and Materials for Integrated Metabolomics
| Item | Function | Platform |
|---|---|---|
| β-Glucuronidase (Type HP-2) | Enzyme to hydrolyze glucuronide-conjugated metabolites, freeing the aglycone for analysis [78]. | GC-MS & LC-MS |
| Methyl-tert-butyldimethylsilyl- N-methyltrifluoroacetamide (MTBSTFA) | Derivatization agent for GC-MS; silanizes polar functional groups (-OH, -COOH, -NH) to increase volatility and thermal stability [78]. | GC-MS |
| Deuterated Internal Standards (e.g., NORD-d5, TEMA-d5) | Corrects for variability in sample preparation, matrix effects, and instrument response; critical for accurate quantification [78]. | GC-MS & LC-MS |
| Solid-Phase Extraction (SPE) Columns | Purify and concentrate analytes from complex biological matrices (e.g., urine, plasma, plant extracts) before instrumental analysis [78]. | GC-MS & LC-MS |
| High-Purity Solvents (ACN, MeOH, Water) | Act as mobile phases for LC separation and for sample preparation/reconstitution. Purity is critical to minimize background noise [78] [3]. | Primarily LC-MS |
| High-Resolution Accurate Mass (HRAM) Instrument | Mass spectrometer capable of exact mass measurement (e.g., Orbitrap, Q-TOF) for confident compound identification and untargeted discovery [113]. | Primarily LC-MS |
| NIST/Wiley EI Mass Spectral Library | Reference database containing fragmentation patterns of hundreds of thousands of compounds for identification based on GC-EI-MS spectra [15] [112]. | GC-MS |
The choice between, or integration of, GC-MS and LC-MS should be driven by specific research questions and sample properties.
Table 5: Decision Guide for Platform Selection
| Criterion | Choose GC-MS if... | Choose LC-MS if... | Prioritize Integration if... |
|---|---|---|---|
| Analyte Properties | Targets are volatile/derivatizable and thermally stable (e.g., essential oils, short-chain acids) [15]. | Targets are polar, ionic, or thermally labile (e.g., peptides, most pharmaceuticals, glycosides) [3]. | The study is truly untargeted, aiming for maximum coverage of unknown metabolites [110]. |
| Identification Need | Reliable identification via universal EI spectral libraries is a top priority [15] [112]. | Structural elucidation via MS/MS and accurate mass is sufficient; authentic standards are available. | Confidence in comprehensive and confident annotation of diverse metabolite classes is critical. |
| Sample Throughput | Derivatization time is acceptable; run times may be longer. | Higher throughput from minimal prep and faster LC cycles is needed [78]. | Throughput can be balanced with depth of information; samples are split for parallel analysis. |
| Budget & Expertise | Capital and operational costs are a major constraint; operational simplicity is valued [15] [3]. | Higher instrument and solvent costs are justified by the required analyte coverage [3]. | The research question demands the highest level of comprehensiveness, justifying the investment in dual-platform analysis. |
Conclusion: For natural product metabolomics and drug discovery research, framing LC-MS versus GC-MS as an either/or proposition is a false dichotomy. The experimental evidence demonstrates that each has distinct and complementary strengths [78] [15]. GC-MS provides robust, library-supported identification for a specific chemical domain, while LC-MS offers expansive coverage of the polar and high molecular-weight metabolome with high sensitivity [2] [14].
Therefore, the most powerful strategy for untargeted profiling is strategic integration. By combining data from both orthogonal platforms, researchers can achieve a more complete and unbiased reconstruction of the metabolic state, leading to more robust biomarker discovery, comprehensive natural product characterization, and a deeper understanding of biological pathways in disease and treatment [111] [110].
In the field of natural product metabolomics, the choice between Liquid Chromatography-Mass Spectrometry (LC-MS) and Gas Chromatography-Mass spectrometry (GC-MS) is fundamental, shaping every subsequent stage of experimental design, analysis, and interpretation [48]. Each platform offers distinct advantages: LC-MS excels in analyzing polar, non-volatile, and thermally labile compounds with minimal pre-analysis derivatization, while GC-MS provides superior separation efficiency and reproducible fragmentation spectra for volatile and thermally stable metabolites [114] [48]. This technical divergence necessitates tailored yet harmonizable validation frameworks to ensure data quality, enable cross-laboratory reproducibility, and support confident biological inference.
The core challenge in validation lies in managing the inherent complexity and dynamic range of natural product metabolomes. A robust framework must integrate standardized experimental protocols, systematic use of reference materials, and sophisticated data correction algorithms to control for technical variance. This guide provides a comparative analysis of validation strategies for LC-MS and GC-MS platforms, grounded in experimental data and contemporary methodologies, to empower researchers in making informed, platform-appropriate decisions for verifying their metabolomic data.
Comparative Analysis of Platform Characteristics and Validation Needs
The foundational differences between LC-MS and GC-MS dictate unique sources of analytical variance and, consequently, distinct focal points for validation protocols.
LC-MS Systems typically employ electrospray ionization (ESI) or atmospheric pressure chemical ionization (APCI), which are sensitive to matrix effects where co-eluting compounds suppress or enhance ionization of analytes. Validation must therefore rigorously assess and compensate for matrix-induced signal variation. Furthermore, modern LC-MS workflows often use data-independent acquisition (DIA) or hybrid modes like Triple Acquisition Mass Spectrometry (TRAM), which require specific validation of quantification accuracy across parallel acquisition cycles [115] [116].
GC-MS Systems traditionally use electron ionization (EI), which generates highly reproducible, library-searchable fragment patterns. The primary validation challenges are associated with sample derivatization (efficiency and completeness) and instrumental drift over time, especially during long sequence runs. The development of novel interfaces like Flame-Induced Atmospheric Pressure Chemical Ionization (FAPCI) for GC-MS also introduces new parameters requiring validation, such as the stability of the micro-flame and ion-molecule reaction efficiency [114] [117].
Table 1: Core Validation Challenges by Analytical Platform
| Validation Aspect | LC-MS (e.g., ESI, APCI) | GC-MS (e.g., EI, FAPCI) |
|---|---|---|
| Primary Source of Variance | Matrix effects, ionization suppression/enhancement, mobile phase composition. | Derivatization efficiency, long-term instrumental drift, column degradation. |
| Quantification Strategy | Relies heavily on stable isotope-labeled internal standards (SIL-IS) for each analyte to correct for matrix effects. | Often uses structural analogs or deuterated standards; can employ internal standards for drift correction across runs [8]. |
| Identification Confidence | High-resolution accurate mass (HRAM) and MS/MS spectral libraries; can be confounded by isobaric compounds. | Library-matchable EI fragment spectra (e.g., NIST); high reproducibility aids identification. |
| Data Acquisition for Validation | DIA provides comprehensive data but requires sophisticated software for deconvolution. Targeted MRM offers high sensitivity. | Full-scan acquisition is standard for untargeted work; SIM mode used for targeted, high-sensitivity analysis. |
| Key Technological Advancements | Hybrid acquisition modes (e.g., TRAM) enabling simultaneous targeted and untargeted analysis in one run [116]. | New ionization sources (e.g., GC-FAPCI) expanding range of analyzable compounds [114] [117]. |
Experimental Protocols for Cross-Platform Method Validation
Protocol for Assessing Long-Term GC-MS Instrument Stability and Drift Correction
Long-term signal drift is a critical challenge for longitudinal studies. The following protocol, adapted from a study on tobacco smoke analysis, provides a systematic approach for monitoring and correcting GC-MS drift over extended periods [8].
- Quality Control (QC) Sample Preparation: Prepare a pooled QC sample that is representative of the entire sample set. For natural products, this could be a pooled extract from multiple representative samples or a carefully crafted mixture of target analytes in a representative matrix.
- Experimental Design: Intersperse the QC sample at regular intervals throughout the entire analytical sequence (e.g., every 5-10 experimental samples). The study in [8] conducted 20 repeated measurements of QCs over 155 days.
- Data Collection and Pre-processing: Analyze the full sequence. For each target compound in the QC injections, extract the peak area or height.
- Drift Modeling and Correction:
- Calculate the median response for each compound across all QC injections to establish a robust "true value" (
X_T,k). - For each QC injection
i, compute a correction factor for compoundk:y_i,k = X_i,k / X_T,k[8]. - Model the correction factor
y_kas a function of batch number (accounting for instrument shutdown/restart) and injection order within a batch using a machine learning algorithm. The cited study found the Random Forest algorithm to provide the most stable and reliable correction model compared to Spline Interpolation or Support Vector Regression [8]. - Apply the derived correction function to the peak areas of the experimental samples based on their batch and injection order.
- Calculate the median response for each compound across all QC injections to establish a robust "true value" (
Protocol for Validating a Hybrid LC-MS/MS Acquisition Method (TRAM)
The TRAM (Triple Acquisition Mass Spectrometry) method unifies full-scan (MS1), data-dependent acquisition (DDA-MS2), and targeted multiple reaction monitoring (MRM-MS2) in a single injection [116]. Its validation must confirm performance parity with dedicated targeted and untargeted methods.
- System Setup: Employ a high-speed quadrupole-time-of-flight (Q-TOF) mass spectrometer capable of rapid cycling. Use a chromatographic method suitable for your metabolite class (e.g., HILIC for polar metabolites) [116].
- Parameter Optimization: Establish a scheduled MRM list with optimized compound-specific parameters (collision energy, declustering potential). Define DDA criteria (e.g., top 30 most intense ions per cycle, dynamic exclusion).
- Validation Experiments:
- Linearity and LOQ: Inject a dilution series of a certified reference material (e.g., NIST SRM 1950 Metabolites in Human Plasma). Assess the linearity and limit of quantification (LOQ) for metabolites quantified via the MRM track. Results should be comparable to a standalone MRM method [116].
- Identification Confidence: Process the DDA data against a spectral library. Compare the number and confidence of metabolite identifications against a standard DDA-only acquisition of the same sample.
- Cross-Validation: For compounds acquired in both MRM and DDA modes, verify that the relative quantification trends across a sample set are consistent between the two data streams.
Protocol for Benchmarking Data Acquisition Modes in LC-MS for PTM Analysis
A study comparing Data-Dependent (DDA) and Data-Independent (DIA) acquisition for histone post-translational modifications provides a template for benchmarking [115].
- Sample Preparation: Use a standardized, complex sample. The cited study used histones extracted from Chinese Hamster Ovary (CHO) cells [115].
- Parallel Acquisitions: Analyze technical replicates of the same sample using different acquisition methods on the same instrument platform. The study compared:
- DDA at MS2 resolutions of 120K, 60K, and 30K.
- DIA with different isolation window schemes (e.g., 20 m/z windows).
- Performance Metrics: Calculate and compare the following for each method:
- Number of Identifications: Unique peptide proteoforms identified.
- Quantification Precision: Coefficient of Variation (CV) for peak areas of identified analytes across replicates. The study found DIA methods could achieve lower CVs [115].
- Spectral Quality: Metrics like Mascot Ion score or Delta score distributions.
- Conclusion Drawing: Determine which method offers the best balance of depth, quantitative precision, and throughput for the specific analytes of interest.
Table 2: Summary of Key Experimental Protocols for Validation
| Protocol Objective | Key Steps | Critical Metrics for Validation | Primary Platform |
|---|---|---|---|
| Correcting Long-Term Drift [8] | 1. Pooled QC sample design.2. Regular interspersion in sequence.3. Algorithmic modeling of drift (e.g., Random Forest). | Reduction in inter-batch CV; clustering of QC samples in PCA after correction. | GC-MS |
| Validating Hybrid Acquisition (TRAM) [116] | 1. Method setup with MRM & DDA tracks.2. Linearity/LOQ with reference material.3. Comparison to standalone methods. | LOQ, linearity (R²), concordance of MRM vs. DDA quantification trends. | LC-MS/MS |
| Benchmarking DDA vs. DIA [115] | 1. Analyze identical samples in multiple modes.2. Compare IDs, precision, scores. | Number of identifications, CV% across replicates, identification confidence scores. | LC-MS/MS (PTM focus) |
The Scientist's Toolkit: Essential Reagents and Reference Materials
A robust validation framework is built upon a foundation of reliable reagents and materials. The following table details key components for natural product metabolomics.
Table 3: Essential Research Reagent Solutions for Metabolomics Validation
| Item | Function in Validation | Example/Note |
|---|---|---|
| Stable Isotope-Labeled Internal Standards (SIL-IS) | Absolute quantification; corrects for matrix effects and recovery losses during extraction. Ideally, one per target analyte. | ¹³C- or ¹⁵N-labeled amino acids, organic acids, lipids. Added at the start of sample preparation [48]. |
| Pooled Quality Control (QC) Sample | Monitors system stability, instrumental drift, and precision over time. | A pool of all study samples or a representative subset. Analyzed repeatedly throughout the batch [8]. |
| Certified Reference Material (CRM) | Validates method accuracy, calibration, and enables cross-laboratory comparison. | NIST SRM 1950 (Metabolites in Human Plasma), plant matrix CRMs [116]. |
| Process Blanks & Solvent Blanks | Identifies background contamination, carryover, and artifacts from solvents or tubes. | Samples containing all reagents but no biological material, processed identically. |
| Derivatization Agents (for GC-MS) | Converts non-volatile metabolites into volatile, thermally stable derivatives. Efficiency must be validated. | MSTFA (N-Methyl-N-(trimethylsilyl)trifluoroacetamide) for silylation; Methoxyamine hydrochloride for oxime formation. |
| Extraction Solvent Systems | Quenches metabolism and extracts metabolites broadly or selectively. Choice significantly impacts results. | Biphasic systems (e.g., methanol/chloroform/water) for global metabolomics; MTBE for lipidomics [48]. |
| Column Performance Test Mixes | Verifies chromatographic separation efficiency and detects column degradation. | Mixtures of standards that elute across the entire gradient, producing known peak shapes and retention times. |
Data Processing and Normalization: Ensuring Comparability
Post-acquisition data processing is a critical component of the validation framework. For untargeted LC-MS or GC-MS data, workflows involve peak picking, alignment, and normalization to mitigate non-biological variance [48]. Normalization strategies include:
- QC-Based: Using the pooled QC sample signal to correct run-order effects [8].
- Internal Standard-Based: Normalizing to one or more spiked SIL-IS.
- Sample-Based: Methods like probabilistic quotient normalization or total useful signal.
For targeted data, integration of peak areas for specific transitions (MRM) or extracted ion chromatograms (EIC) is followed by normalization to the corresponding SIL-IS peak area, which is the gold standard for achieving accurate concentration data [116].
Visualization of Validation Workflows and Decision Pathways
Validation Workflow for Metabolomics Data
Platform Selection and Validation Focus Decision Tree
Effective validation in natural product metabolomics is not a one-size-fits-all process but a platform-informed strategy. For GC-MS, the framework must prioritize controlling long-term instrumental drift and ensuring derivatization reproducibility, leveraging algorithmic correction and robust QC placement [8]. For LC-MS, the focus shifts to managing matrix effects through comprehensive SIL-IS use and validating the performance of complex acquisition modes like DIA or TRAM [115] [116].
The convergence of these frameworks lies in the universal application of certified reference materials, pooled QCs, and process blanks. As hybrid and novel ionization techniques evolve, validation protocols must adapt accordingly. By adopting the comparative guidelines and experimental protocols outlined here, researchers can generate metabolomic data that is not only analytically sound but also comparable across studies and platforms, thereby strengthening the collective understanding of natural product biochemistry.
The comprehensive analysis of natural products presents a formidable challenge to modern metabolomics. These complex samples contain hundreds to thousands of metabolites with vast chemical diversity, spanning volatile essential oils to large, polar glycosides. For decades, the field has relied on two cornerstone separation techniques coupled with mass spectrometry: gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-mass spectrometry (LC-MS). Each platform has distinct advantages and inherent limitations, making the choice between them a fundamental first step in experimental design [15]. However, even these powerful techniques can struggle with definitive identification in highly complex matrices, where co-eluting isomers and "known unknown" compounds are prevalent.
This is where a transformative technological synergy emerges. The integration of ion mobility spectrometry (IMS) adds a rapid, gas-phase separation dimension based on an ion's size and shape, providing a reproducible collision cross-section (CCS) value that serves as a powerful molecular descriptor [118]. Concurrently, machine learning (ML) algorithms are revolutionizing data processing, moving beyond simple library matching to predict properties, classify samples, and uncover hidden patterns in high-dimensional data [119]. This guide objectively compares the performance of traditional GC-MS and LC-MS within natural product research and demonstrates how the integration of IMS and ML is pushing beyond traditional analytical boundaries, offering researchers unprecedented confidence in metabolite identification and biological interpretation.
Methodology Comparison: LC-MS vs. GC-MS for Natural Products
The choice between LC-MS and GC-MS is not a matter of superiority but of appropriate application, dictated by the physicochemical properties of the target analytes and the research question. The following table provides a foundational comparison for researchers in natural product metabolomics.
Table 1: Core Comparison of GC-MS and LC-MS in Metabolomics [15]
| Criterion | GC-MS | LC-MS |
|---|---|---|
| Ideal Analytic Profile | Volatile and thermally stable compounds; typically < ~500 Da. | Polar, ionic, and thermally labile compounds; wide range from small metabolites to large biomolecules (>10 kDa). |
| Primary Ionization | Electron Ionization (EI) - a "hard" method producing extensive, reproducible fragmentation. | Electrospray Ionization (ESI) or Atmospheric Pressure Chemical Ionization (APCI) - "soft" methods often preserving the molecular ion. |
| Separation Basis | Volatility and interaction with a stationary phase in a heated column. | Polarity, hydrophobicity, and affinity in a liquid-phase column at ambient temperature. |
| Key Strength | Excellent chromatographic resolution for structural isomers; highly reproducible retention times and EI spectra; mature, extensive spectral libraries (e.g., NIST) for confident identification. | Broad coverage of metabolite space, especially polar bioactive compounds (e.g., flavonoids, saponins); high sensitivity in targeted bioanalysis; minimal need for derivatization. |
| Major Limitation | Requires volatility, often necessitating derivatization for polar compounds, which adds steps and can complicate quantification. | Susceptible to matrix effects that can suppress or enhance ionization; less standardized fragmentation patterns and smaller spectral libraries compared to EI. |
| Typical Natural Product Applications | Analysis of essential oils, mono- and sesquiterpenes, fatty acids, certain alkaloids (after derivatization). | Analysis of phenolics, flavonoids, glycosides, polar lipids, most alkaloids, peptides. |
Advancing the Platform: The Integrated Role of IMS and ML
To address the limitations of both GC-MS and LC-MS—particularly challenges in separating isobaric/isomeric compounds and confidently annotating unknown features—IMS and ML are being integrated as complementary technologies.
Table 2: Performance Enhancements from IMS and ML Integration
| Technology | Primary Role | Key Performance Metric & Impact | Supporting Data |
|---|---|---|---|
| Ion Mobility Spectrometry (IMS) | Adds a rapid (ms), orthogonal separation dimension based on an ion's collision cross-section (CCS) in a buffer gas. | Increased Identification Confidence: Using LC-IMS-MS, peak matching with mass, retention time, and CCS value reduced the false discovery rate (FDR) by ~30% compared to using only mass and retention time [118]. Provides a reproducible physicochemical descriptor for "known unknowns." | Study on human serum peptides demonstrated enhanced confidence in identifications [118]. |
| Machine Learning (ML) | Processes high-dimensional data for pattern recognition, predictive modeling, and automated classification. | Improved Classification Accuracy: In fire debris analysis using HS-GC-IMS, ML models (SVM, Random Forest) detected ignitable liquid residues with 100% accuracy [120]. Enables automated, high-throughput analysis of complex samples. | ML applied to IMS data for forensic classification [120]. |
| ML in LC-MS Non-Targeted Analysis (NTA) | Manages complexity in untargeted workflows: peak alignment, feature selection, and predicting properties like retention time (RT) and CCS. | Enhanced Efficiency & Discovery: ML models bridge the gap between spectra and molecular structures, accelerating metabolite annotation where libraries are lacking [119]. Reduces manual data interrogation time significantly. | Review highlights ML's role in LC-MS NTA for metabolomics and environmental science [119]. |
The integration of these technologies creates a more powerful analytical workflow. IMS provides an additional, orthogonal data point (CCS) that improves the specificity of compound matching. ML algorithms can then leverage this richer, multi-dimensional data (m/z, RT, CCS, fragmentation pattern) to build more accurate predictive models for compound identification and classification, especially for isomers and novel compounds not present in standard libraries.
Experimental Protocols for Integrated Analysis
A robust metabolomics workflow is critical for generating reliable data, especially when integrating advanced technologies like IMS and ML [1].
Sample Preparation & Metabolite Extraction
- Sample Collection & Quenching: Use rapid freezing in liquid nitrogen or cold organic solvents (e.g., -40°C methanol) to instantly quench metabolic activity in tissues or cells, preserving the in vivo metabolite profile [1].
- Extraction: For broad-coverage untargeted analysis of natural products, a biphasic solvent system is often employed. A common method uses a methanol/chloroform/water mixture (e.g., 2:1:1 ratio). This simultaneously extracts polar metabolites (into the methanol/water layer) and non-polar lipids (into the chloroform layer) [1].
- Quality Control (QC): Incorporate internal standards (stable isotope-labeled analogs of key metabolites) during extraction to correct for losses and variability. Consistently run pooled QC samples throughout the analytical sequence to monitor instrument stability [1].
Instrumental Analysis: LC-IMS-MS Workflow
- Chromatography: For LC-MS, use a C18 reversed-phase column with a water-acetonitrile gradient (both acidified with 0.1% formic acid) for broad separation. For complex natural product extracts, ultra-high-performance liquid chromatography (UHPLC) is recommended for superior resolution [2].
- Ion Mobility Separation: Post-LC separation, ions are pulsed into a drift tube IMS cell filled with an inert buffer gas (e.g., nitrogen). Under a weak electric field, ions separate based on their size-to-charge ratio, yielding a drift time that is converted into a collision cross-section (CCS) value—a reproducible identifier [118] [121].
- Mass Spectrometry Detection: A high-resolution mass spectrometer (e.g., Q-TOF) is used after the IMS cell. This configuration provides data in four dimensions: retention time, drift time, mass-to-charge ratio (m/z), and intensity.
Data Processing & Machine Learning Analysis
- Feature Extraction: Use specialized software (e.g., Decon2LS, MS-DIAL) to deconvolute raw data, aligning peaks across samples and detecting features defined by m/z, RT, and CCS [118].
- ML Model Application:
- For Classification: Use supervised ML models like Random Forest (RF) or Support Vector Machines (SVM). The model is trained on a labeled dataset (e.g., samples from different plant species or treatment groups) using the multidimensional features (RT, CCS, m/z intensities) as input. The trained model can then classify new samples and identify the most discriminatory features (potential biomarkers) [120] [119].
- For Prediction: Use algorithms to predict molecular properties. For example, a model can be trained to predict a metabolite's CCS value from its molecular fingerprint. A significant deviation between the measured and predicted CCS for a putative identity can flag a potentially incorrect annotation [119].
- Visualization & Interpretation: Employ multivariate statistics (PCA, PLS-DA) and ML-driven visualizations (cluster heatmaps, volcano plots) to interpret results. Network analysis can connect discriminative metabolites to biological pathways [122] [123].
Integrated LC-IMS-MS Workflow with ML
The Scientist's Toolkit: Essential Research Reagents & Materials
Table 3: Key Reagents and Materials for Natural Product Metabolomics Workflows
| Item | Function & Rationale | Key Considerations |
|---|---|---|
| Quenching Solvent (e.g., Cold Methanol, Liquid N₂) | Instantly halts enzymatic activity to "freeze" the metabolic state at the moment of sampling [1]. | Speed is critical. For cells, cold (-40°C to -80°C) aqueous methanol (60-80%) is commonly used. |
| Biphasic Extraction Solvents (Methanol/Chloroform/Water) | Provides comprehensive coverage by extracting both polar and non-polar metabolites simultaneously [1]. | Ratios can be optimized (e.g., 2:1:1 or 1:2:0.8). Chloroform handles non-polar lipids, methanol/water extracts polar compounds. |
| Stable Isotope-Labeled Internal Standards | Added at extraction start to correct for analyte loss and matrix effects; essential for reliable quantification [1]. | Should cover various chemical classes. Examples: ¹³C-labeled amino acids, ²H-labeled lipids. |
| LC-MS Grade Solvents & Additives | Used for mobile phases to ensure low background noise, prevent ion source contamination, and achieve reproducible chromatography [2]. | Acetonitrile and methanol (LC-MS grade) with additives like 0.1% formic acid (for positive mode) or ammonium acetate (for negative mode). |
| Derivatization Reagents (for GC-MS) | Chemical modification (e.g., silylation with MSTFA) to increase volatility and thermal stability of polar metabolites for GC-MS analysis [15]. | Adds a sample preparation step; can generate multiple derivatives per compound. Must be performed under anhydrous conditions. |
| IMS Drift Gas (e.g., Pure N₂ or CO₂) | Inert gas filling the IMS drift tube; ions separate via collisions with this gas, defining the CCS measurement [118] [121]. | Must be ultra-pure and dry. Humidity can cluster with ions, altering drift times and reducing reproducibility. |
| QC Pooled Sample | A homogeneous mixture of all study samples, injected repeatedly throughout the analytical sequence [1]. | Monitors instrument performance (retention time stability, signal intensity, mass accuracy) and is essential for data normalization. |
The metabolomic analysis of natural products no longer requires a strict choice between GC-MS and LC-MS but rather a strategic understanding of their complementary strengths. GC-MS remains unmatched for the resolved analysis of volatile compounds with robust library-based identification, while LC-MS is indispensable for profiling the vast space of polar, bioactive molecules.
The true frontier, however, lies in seamlessly integrating these platforms with ion mobility spectrometry (IMS) and machine learning (ML). IMS delivers an orthogonal separation and a reproducible molecular descriptor (CCS) that dramatically increases confidence in distinguishing isomers and annotating unknowns. ML transforms the resulting high-dimensional data from a processing challenge into a discovery opportunity, enabling automated classification, predictive modeling, and the extraction of biologically meaningful patterns. For researchers and drug development professionals, this synergistic approach—moving beyond traditional analysis—offers a more powerful, informative, and efficient path to unlocking the complex chemistry of natural products, from quality control to novel biomarker discovery.
Conclusion
LC-MS and GC-MS are not competing but profoundly complementary pillars of natural product metabolomics. GC-MS offers excellent sensitivity, reproducibility, and robust compound identification via standardized libraries for volatile and derivatizable compounds[citation:1][citation:4]. In contrast, LC-MS provides unparalleled breadth in analyzing semi-polar and non-volatile metabolites, including complex lipids, without the need for derivatization[citation:1][citation:8]. The choice between them is primarily dictated by the physicochemical properties of the target metabolite classes. For the most comprehensive systems-level view—essential for deconvoluting the complex chemistry of natural sources—an integrated multiplatform approach is increasingly considered best practice[citation:5][citation:7]. Future directions point toward the tighter coupling of these platforms with advanced computational tools like machine learning for structure elucidation and the development of high-throughput, automated workflows[citation:7][citation:9]. By making informed, strategic choices between or combining GC-MS and LC-MS, researchers can significantly enhance the depth and reliability of their findings, accelerating the discovery and validation of bioactive natural products for biomedical and clinical applications.
References
- 1. Mass-spectrometry based metabolomics: an overview of ... [https://proteomesci.biomedcentral.com/articles/10.1186/s12953-025-00241-8]
- 2. Recent Advances in Liquid Chromatography–Mass ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12204688/]
- 3. LCMS vs. GCMS: When to Choose Each for Optimal ... [https://emerypharma.com/blog/lcms-vs-gcms-when-to-choose-each-for-optimal-results-in-your-analytical-chemistry/]
- 4. Metabolomics by Gas Chromatography-Mass Spectrometry [https://pmc.ncbi.nlm.nih.gov/articles/PMC4829120/]
- 5. Mass Spectrometry for Natural Products Research [https://www.chromatographyonline.com/view/mass-spectrometry-natural-products-research-challenges-pitfalls-and-opportunities]
- 6. A Multi-omics approach combining GC-MS, LC-MS, and FT ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967325004844]
- 7. Optimization of GC-MS run time for untargeted metabolomics [https://www.sciencedirect.com/science/article/abs/pii/S0731708525004091]
- 8. Correction of gas chromatography–mass spectrometry long ... [https://www.nature.com/articles/s41598-025-24794-y]
- 9. MetaboAnalystR 4.0: a unified LC-MS workflow for global ... [https://www.nature.com/articles/s41467-024-48009-6]
- 10. The Cinderella story of metabolic profiling [https://pmc.ncbi.nlm.nih.gov/articles/PMC1626538/]
- 11. GC-MS-Based Metabolomics | SpringerLink [https://link.springer.com/protocol/10.1007/978-1-59745-463-6_15]
- 12. Principles and Differences between GC-MS and LC-MS [https://metabolomics.creative-proteomics.com/resource/principles-and-differences-between-gc-ms-and-lc-ms.htm]
- 13. GC-MS Metabolomics Analysis [https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/mass-spectrometry-learning-center/mass-spectrometry-applications-area/metabolomics-mass-spectrometry/practical-guide-metabolomics/gc-ms-metabolomics-analysis.html]
- 14. LC-MS VS GC-MS: What's the Difference [https://www.metwarebio.com/lc-ms-vs-gc-ms-whats-the-difference/]
- 15. GC-MS vs LC-MS: How to Choose for Metabolomics ... [https://www.arome-science.com/metabolomics/gc-ms-vs-lc-ms-how-to-choose-for-metabolomics-research/]
- 16. NMR and GC-MS based metabolic profiling, total phenolic ... [https://www.nature.com/articles/s41598-025-26621-w]
- 17. Nutritional Characterization and Metabolic Profiling of ... [https://pubmed.ncbi.nlm.nih.gov/40377812/]
- 18. in the Context of Plant Metabolomics Research... Natural Products [https://pmc.ncbi.nlm.nih.gov/articles/PMC7023240/]
- 19. advances in metabolomics for natural product drug discovery [https://www.sciencedirect.com/science/article/pii/S095816692500117X?via%3Dihub]
- 20. The Utility of Metabolomics in Natural Product and Biomarker ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4475408/]
- 21. Mass spectrometry-based metabolomic as a powerful tool to ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12067679/]
- 22. The Role of LC-MS in Profiling Bioactive Compounds from ... [https://www.mdpi.com/2223-7747/14/15/2284]
- 23. Gas Chromatography/ Mass Spectrometry Fundamentals [https://www.agilent.com/en/product/gas-chromatography-mass-spectrometry-gc-ms/gcms-fundamentals?srsltid=AfmBOoqx9U0QYa4O3amnZaPnEEoJLIvdbDkzxTHGNqZXUIA_9qkmX-OY]
- 24. Gas Chromatography Mass Spectrometry (GC-MS) ... [https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/mass-spectrometry-learning-center/gas-chromatography-mass-spectrometry-gc-ms-information.html]
- 25. Liquid Chromatography/ Mass Spectrometry Fundamentals [https://www.agilent.com/en/product/liquid-chromatography-mass-spectrometry-lc-ms/lcms-fundamentals?srsltid=AfmBOopkGeKXXDF8lX1uygNbyZLECQ_mFRAqNwokkS0i0zQu7fckRxPO]
- 26. Liquid chromatography–mass spectrometry [https://en.wikipedia.org/wiki/Liquid_chromatography%E2%80%93mass_spectrometry]
- 27. Advances in Dual-Column Chromatography for ... [https://www.sciencedirect.com/science/article/pii/S2772391725000647]
- 28. GC/MS and LC/MS serum metabolomic analysis of ... [https://www.nature.com/articles/s41598-024-52137-w]
- 29. Metabolomics based on GC-MS combined with ... [https://www.sciencedirect.com/science/article/abs/pii/S0956713524006935]
- 30. LC-MS-based metabolomics: targeted and untargeted ... [https://www.sciencedirect.com/science/article/pii/B9780443337109000078]
- 31. LC-MS-based metabolomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC3699692/]
- 32. Integration of GC–MS and LC–MS for untargeted ... [https://www.sciencedirect.com/science/article/abs/pii/S0731708520313959]
- 33. Principles and Applications of Liquid Chromatography ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2643089/]
- 34. Metabolomics Sample Preparation [https://www.organomation.com/metabolomics-sample-preparation]
- 35. Techniques for extraction and isolation of natural products [https://pmc.ncbi.nlm.nih.gov/articles/PMC5905184/]
- 36. Contemporary methods for the extraction and isolation of ... [https://bmcchem.biomedcentral.com/articles/10.1186/s13065-023-00960-z]
- 37. Solvent Extraction Techniques [https://www.organomation.com/solvent-extraction-techniques]
- 38. Comparing the Chemical Profiles of Plant-Based and ... [https://www.chromatographyonline.com/view/comparing-the-chemical-profiles-of-plant-based-and-traditional-meats-using-gc-ms-based-metabolomics]
- 39. SPE Offers Multiple Advantages Over LCC for Hydrocarbon ... [https://www.chromatographyonline.com/view/spe-offers-multiple-advantages-over-lcc-for-hydrocarbon-analysis-by-gc-ms]
- 40. Liquid and gas-chromatography-mass spectrometry ... [https://www.sciencedirect.com/science/article/pii/S0021967325000779]
- 41. Application of Metabolomics to Quality Control of Natural ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5685424/]
- 42. Accurate and robust method for plant volatilome analysis ... [https://www.sciencedirect.com/science/article/pii/S2666831925001791]
- 43. Application of gas chromatography-mass spectrometry ... [https://www.sciencedirect.com/science/article/abs/pii/S1389172322000111]
- 44. Analyzing Non-Volatile Compounds with GC-MS: A Guide [https://www.hplcvials.com/faq/analyzing-non-volatile-compounds-with-gc-ms-a-guide.html]
- 45. Derivatization Methods in GC and GC/MS [https://www.intechopen.com/chapters/64643]
- 46. Comparison of derivatization and chromatographic ... [https://pubmed.ncbi.nlm.nih.gov/20371216/]
- 47. Chemometrics comparison of gas with chromatography ... mass [https://pubmed.ncbi.nlm.nih.gov/29485703/]
- 48. Mass-spectrometry based metabolomics - PubMed Central - NIH [https://pmc.ncbi.nlm.nih.gov/articles/PMC12105183/]
- 49. – Gas ( Chromatography – Mass ) with Cold... Spectrometry GC MS [https://www.spectroscopyonline.com/view/gas-chromatography-mass-spectrometry-gc-ms-with-cold-electron-ionization-ei-bridging-the-gap-between-gc-ms-and-lc-ms]
- 50. Performance comparison of electrospray ionization and ... [https://pubmed.ncbi.nlm.nih.gov/27935129/]
- 51. Understanding the atmospheric pressure ionization of ... [https://www.sciencedirect.com/science/article/abs/pii/S0048969716312177]
- 52. Efficient and Wide Chemical-Space Ionization of Organic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12489893/]
- 53. Comparison of ESI– and APCI–LC–MS/MS methods [https://pmc.ncbi.nlm.nih.gov/articles/PMC5762935/]
- 54. of Comparison - gas and chromatography ... mass spectrometry gas [https://pubmed.ncbi.nlm.nih.gov/19609395/]
- 55. of different fast Comparison – gas ... chromatography mass [https://pubs.rsc.org/en/content/articlelanding/2024/ay/d4ay00858h]
- 56. GC-MS vs LC-MS [https://resolvemass.ca/gc-ms-vs-lc-ms/]
- 57. GC-MS Fingerprinting Combined with Chemical Pattern ... [https://pubmed.ncbi.nlm.nih.gov/38067553/]
- 58. Tips and tricks for LC–MS-based metabolomics and ... [https://www.sciencedirect.com/science/article/pii/S0165993624004230]
- 59. North America Mass Spectrometry Market Report 2025 ... [https://finance.yahoo.com/news/north-america-mass-spectrometry-market-091100776.html]
- 60. Top 10 Companies in Mass Spectrometry Market (2025– ... [https://www.sphericalinsights.com/blogs/top-10-companies-in-mass-spectrometry-market-2025-2035-competitive-analysis-and-forecast]
- 61. The Difference Between GC/MS and LC/MS Systems [https://gentechscientific.com/the-difference-between-gc-ms-and-lc-ms-systems/]
- 62. The Difference Between GC/MS and LC/MS Systems [https://conquerscientific.com/the-difference-between-gc-ms-and-lc-ms-systems/]
- 63. Perspectives of Quantitative GC-MS, LC-MS, and ICP-MS in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11641993/]
- 64. LC-MS/MS-Based Diagnostics Market Trends and ... [https://www.globenewswire.com/news-release/2025/10/07/3162438/0/en/LC-MS-MS-Based-Diagnostics-Market-Trends-and-Competitive-Landscape-Report-2024-2034.html]
- 65. Harnessing Mass Spectrometry for Biomarker Discovery in ... [https://blog.crownbio.com/biomarker-and-mass-spectrometry-aml]
- 66. Ion Suppression: A Major Concern in Mass Spectrometry [https://www.chromatographyonline.com/view/ion-suppression-major-concern-mass-spectrometry]
- 67. Systematic Assessment of Matrix Effect, Recovery, and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12613129/]
- 68. Overview of Mass Spectrometry-Based Metabolomics: Opportunities and Challenges - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC4336784/]
- 69. Matrix Effects in Untargeted LC-MS Metabolomics [https://www.sciencedirect.com/science/article/pii/S0021967325008520]
- 70. LC-MS/MS 2025 Trends and Forecasts 2033 [https://www.archivemarketresearch.com/reports/lc-msms-287243]
- 71. Liquid Chromatography-Mass Spectrometer (LC-MS) Market [https://dataintelo.com/report/liquid-chromatography-mass-spectrometer-lc-ms-market]
- 72. New HPLC, MS, and CDS Products from 2024–2025 [https://www.chromatographyonline.com/view/new-hplc-ms-and-cds-products-from-2024-2025-a-brief-review]
- 73. Optimization of derivatization-gas chromatography/mass ... [https://www.sciencedirect.com/science/article/abs/pii/S0308814625024033]
- 74. Comparison of gas chromatography–mass spectrometry ... [https://www.sciencedirect.com/science/article/abs/pii/S0021967303002176]
- 75. Optimization of a GC-MS Injection-Port Derivatization ... [https://pubmed.ncbi.nlm.nih.gov/36180971/]
- 76. Trivialities in metabolomics: Artifacts in extraction and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9493124/]
- 77. The Origin and Implications of Artifact Ions in Bioanalytical ... [https://www.chromatographyonline.com/view/the-origin-and-implications-of-artifact-ions-in-bioanalytical-lc-ms]
- 78. Comparison of LC–MS-MS and GC–MS Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4885916/]
- 79. Advancing Organic Chemistry Using High‐Throughput ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12462754/]
- 80. High Throughput Liquid and Gas Chromatography ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3803150/]
- 81. GC-MS and HPLC chemical profile, antioxidant, anti ... [https://www.nature.com/articles/s41598-025-12233-x]
- 82. Automated Workflow for High-Throughput LC-MS ... [https://pubmed.ncbi.nlm.nih.gov/40725248/]
- 83. Automated Workflow for High-Throughput LC–MS ... [https://www.mdpi.com/1422-0067/26/14/6999]
- 84. A fully automated, high-throughput electro-extraction and ... [https://www.sciencedirect.com/science/article/pii/S000326702500618X]
- 85. LC–MS-MS Total Drug Analysis of Biological Samples ... [https://www.chromatographyonline.com/view/lc-ms-ms-total-drug-analysis-biological-samples-using-high-throughput-protein-precipitation-method]
- 86. High-throughput analytical methods employing ... [https://www.sciencedirect.com/science/article/pii/S2772582023000451]
- 87. Analytical Solutions for High Throughput ADME Studies [https://www.waters.com/nextgen/us/en/library/application-notes/2024/analytical-solutions-for-high-throughput-adme-studies.html?srsltid=AfmBOoro0ISTqLQRZD-IcZFqVA9DjLiJFYN-IfVMYeuIPqVK0AZe_21V]
- 88. mini review article untargeted metabolomics: an emerging ... [https://www.sciencedirect.com/science/article/pii/S2001037014600507]
- 89. Metabolomics in Natural Product Analysis [https://www.chromatographyonline.com/view/metabolomics-natural-product-analysis]
- 90. Design of Experiments | Statistics Knowledge Portal [https://www.jmp.com/en/statistics-knowledge-portal/design-of-experiments]
- 91. Design of Experiments in metabolomics-related studies [https://www.sciencedirect.com/science/article/pii/S0731708518317229]
- 92. Optimization through classical design of experiments (DOE) [https://www.sciencedirect.com/science/article/pii/S2352710225001676]
- 93. Designing optimal experiments in metabolomics [https://pubmed.ncbi.nlm.nih.gov/38941008/]
- 94. A comprehensive review on computational metabolomics [https://www.sciencedirect.com/science/article/pii/S2001037025002806]
- 95. Metabolomics Quality Control, Reproducibility & Validation [https://www.arome-science.com/metabolomics-faqs/metabolomics-quality-control-reproducibility-method-validation-guide/]
- 96. IS Responses in LC-MS/MS Bioanalysis [https://www.biopharmaservices.com/blog/bioanalysis-internal-standard-responses-in-lc-ms-ms-friend-or-foe/]
- 97. When Should an Internal Standard be Used? [https://www.chromatographyonline.com/view/when-should-internal-standard-be-used-0]
- 98. Applications of liquid chromatography-mass spectrometry ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9669074/]
- 99. Comparison of GC-MS and GC×GC-MS in the Analysis ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4849136/]
- 100. Comparison of LC–MS and GC–MS for the Analysis ... [https://www.spectroscopyonline.com/view/comparison-lc-ms-and-gc-ms-analysis-pharmaceuticals-and-personal-care-products-surface-water-and-tre]
- 101. Orbitrap LC-MS Comparison Chart [https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-systems/orbitrap-lc-ms/orbitrap-lc-ms-comparison-chart.html]
- 102. HS-SPME-GC–MS Profiling of Volatile Organic ... [https://www.mdpi.com/2311-7524/10/3/257]
- 103. Comparative analysis of continuous similarity measures for ... [https://chemrxiv.org/engage/chemrxiv/article-details/6699766801103d79c55cc41f]
- 104. Metabolic Profiling of Oxidized Lipid-Derived Volatiles in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3967009/]
- 105. Towards high-throughput metabolomics using ultrahigh ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2600444/]
- 106. Targeted vs Untargeted Metabolomics [https://www.metabolon.com/blog/targeted-vs-untargeted-metabolomics/]
- 107. Targeted vs Untargeted Metabolomics [https://metabolomics.creative-proteomics.com/resource/comparative-analysis-targeted-vs-untargeted-metabolomics.htm]
- 108. Targeted vs Untargeted vs Semi-Targeted Metabolomics [https://www.arome-science.com/metabolomics/targeted-vs-untargeted-vs-semi-targeted-metabolomics/]
- 109. Targeted vs Untargeted vs Widely-targeted Metabolomics [https://www.metwarebio.com/targeted-vs-untargeted-metabolomics/]
- 110. Integration of GC–MS and LC–MS for untargeted ... [https://www.sciencedirect.com/science/article/pii/S0731708520313959]
- 111. Integration of GC-MS and LC-MS for untargeted ... [https://pubmed.ncbi.nlm.nih.gov/32814263/]
- 112. Untargeted Metabolomics Workflows [https://www.thermofisher.com/us/en/home/industrial/mass-spectrometry/mass-spectrometry-learning-center/mass-spectrometry-applications-area/metabolomics-mass-spectrometry/metabolomics-workflows/untargeted-metabolomics-workflows.html]
- 113. Orbitrap LC-MS Comparison Chart [https://www.thermofisher.com/tr/en/home/industrial/mass-spectrometry/liquid-chromatography-mass-spectrometry-lc-ms/lc-ms-systems/orbitrap-lc-ms/orbitrap-lc-ms-comparison-chart.html]
- 114. Gas chromatography combined with flame-induced ... [https://www.sciencedirect.com/science/article/abs/pii/S0003267022001829]
- 115. Comparison of data‐acquisition methods for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6519233/]
- 116. Triple acquisition mass spectrometry (TRAM) combining ... [https://www.sciencedirect.com/science/article/pii/S0003267024011152]
- 117. Gas chromatography combined with flame-induced ... [https://pubmed.ncbi.nlm.nih.gov/35256141/]
- 118. Increasing Confidence of LC-MS Identifications by Utilizing ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4114398/]
- 119. Application of machine learning in LC-MS-based non- ... [https://www.sciencedirect.com/science/article/abs/pii/S0165993625001116]
- 120. Machine learning approaches over ion mobility spectra for ... [https://www.sciencedirect.com/science/article/pii/S2666831922000431]
- 121. Gas Chromatography and Ion Mobility Spectrometry [https://www.chromatographyonline.com/view/gas-chromatography-and-ion-mobility-spectrometry-a-perfect-match-]
- 122. Visualizing Metabolomics Data: Graphical Insights [https://metabolomics.creative-proteomics.com/resource/visualizing-metabolomics-data-graphical-insights.htm]
- 123. Effective data visualization strategies in untargeted ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11610048/]